

 1592
1592
要回答这些问题,激发态计算(Excited-State Calculation)就成了关键。它是连接分子结构与光谱性质的桥梁,使理论化学家能够用计算机“看到”分子在光照下的反应。
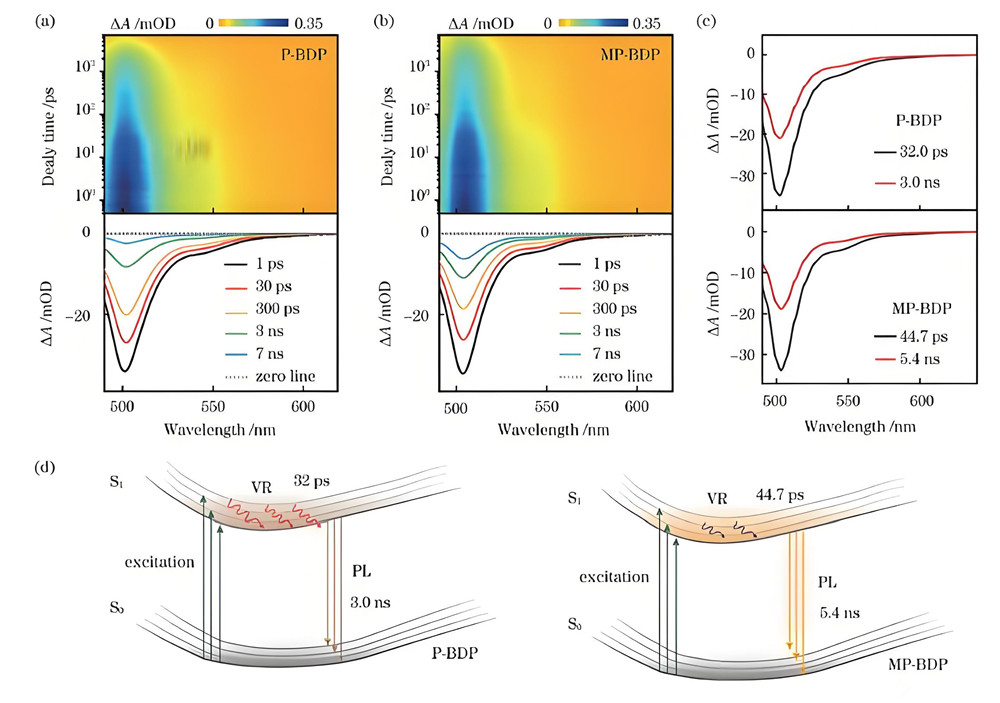
激发态计算是量子化学中研究分子吸收能量后电子跃迁至较高能级状态的重要方法,其核心在于分析激发态的能量、结构、光谱性质及反应活性。
如今,Gaussian(高斯)软件已经成为最常用的量子化学工具之一,其中的激发态计算模块能帮助研究者预测吸收光谱、分析发光性质,甚至揭示光化学反应机理。
本文将带你了解什么是激发态计算,它是怎么做的,有什么应用与局限。
一、什么是激发态计算
激发态计算的核心目标,就是通过量子化学方法预测:
激发态的能量(电子跃迁需要的能量,即光吸收波长);
激发态的类型(单重态、三重态等自旋多重度);
光吸收或发射的强度(振子强度);
激发电子与空轨道的分布(跃迁特征)。
二、激发态计算方法
TD-DFT:在密度泛函理论(DFT)基础上,引入时间依赖项,通过求解分子对微小外场扰动的响应,得到电子从基态跃迁到激发态时的能量差。它的优点是计算速度快、精度较好,尤其适用于中小分子的吸收光谱模拟。
CIS(单激发组态相互作用):是最基础的激发态波函数方法,虽然精度一般,但计算代价低,适合教学与入门理解。
EOM-CCSD(方程-运动耦合簇):属于高精度波函数方法,能很好地处理单激发主导的体系,但计算成本较高,通常用于小分子或验证性计算。
在实际研究中,研究者通常会:
三、激发态计算的应用
光谱模拟与解析:通过理论计算吸收峰位置与强度,帮助解释实验 UV-Vis 光谱或拉曼光谱。
荧光与磷光研究:揭示发光材料(如OLED分子)的激发态性质,预测发光波长与效率。
光催化机理:研究光催化剂在激发态下的电子跃迁路径与反应通道。
光敏分子设计:用于开发染料敏化太阳能电池、光响应分子开关等新材料。
四、优缺点与注意事项

泛函与基组选择会显著影响结果。例如,电荷转移态常需长程修正泛函(如 CAM-B3LYP)与含弥散函数的基组(如 6-31+G*)。
激发态优化过程中可能出现态交叉或轨道重排,需要结合轨道分析确认是否跟踪到正确的激发态。
对于自旋多重度不同的态,应设置合适的自旋限制条件,以避免得到错误解。
五、总结
在实践中,TD-DFT 是最常用的主力方法,能够快速预测光谱并揭示激发态的本质。对于要求更高的研究,则可辅以 EOM-CCSD 等精确方法。可以说,激发态计算让我们不再只是“看见”光谱,而是让我们得以在理论层面去探索分子世界。


