

 2322
2322
DFT 中的自洽计算(SCF)与非自洽计算(Non-SCF) ,核心区别在于:是否通过迭代更新电子密度以满足自洽条件,二者的计算目的、流程和参数设置截然不同。
一、 核心定义与本质区别
1、自洽计算(SCF)
自洽计算的核心目标是求解基态电子密度。
它基于 Kohn-Sham 方程,通过迭代循环实现:先假设一个初始电子密度,计算对应的势函数,求解单电子方程得到波函数,再由波函数重新计算电子密度;重复此过程,直到两次迭代的电子密度(或总能量)的差异小于设定阈值,达到自洽收敛。
收敛后得到的电子密度、总能量、电荷密度等,是体系的基态性质。
2、非自洽计算(Non-SCF)
非自洽计算不更新电子密度,而是固定电子密度(通常取自 SCF 计算的收敛结果),在此基础上求解 Kohn-Sham 方程,计算特定需求的物理量。
因为电子密度不再迭代更新,所以计算速度远快于 SCF,且不需要满足自洽收敛条件。
二、关键差异对比
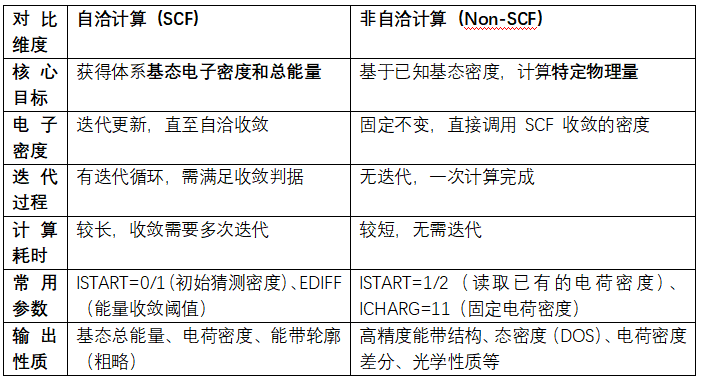
三、 典型应用场景
1、自洽计算的应用
优化晶胞参数和原子位置(结构弛豫,IBRION≠0时默认包含 SCF);
计算体系的基态总能量、形成能、结合能;
获得收敛的电荷密度,为后续 Non-SCF 计算提供输入。
2、非自洽计算的应用
高精度能带结构计算:先通过 SCF 得到基态密度,再选取高对称 k 点路径,用 Non-SCF 计算能带(需设置KPOINTS为高对称路径);
态密度(DOS)计算:使用密集的 k 点网格,固定电荷密度计算 DOS,提高精度;
电荷密度差分、局域态密度(LDOS):基于固定密度分析电子分布的变化;
光学性质、介电函数等进阶性质的计算。
四、 二者的关联
在 VASP 的实际计算中,Non-SCF 计算几乎总是依赖于 SCF 计算的结果:
1、先进行 SCF 计算,得到收敛的电荷密度文件(CHGCAR)和波函数文件(WAVECAR);
2、保持体系结构不变,修改INCAR参数(如设置ICHARG=11)、KPOINTS文件(如高对称路径),执行 Non-SCF 计算,调用CHGCAR中的电荷密度进行后续求解。



