

 7700
7700
在第一性原理计算软件VASP中,能带结构(Band Structure)是研究材料电子性质的核心内容之一。通过计算材料的能带图,我们可以直观了解电子在晶体中的能量分布,从而判断材料的导电类型:金属、半导体、绝缘体或半金属等。本文将详细介绍VASP中能带结构的计算流程、解读方法,以及基于能带结构的材料分类。
一、什么是能带结构?
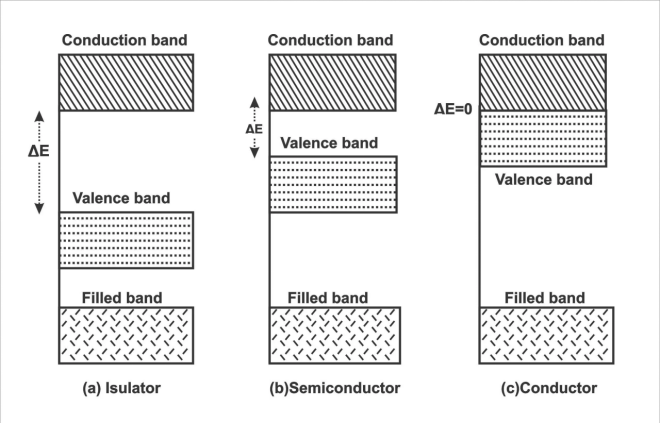
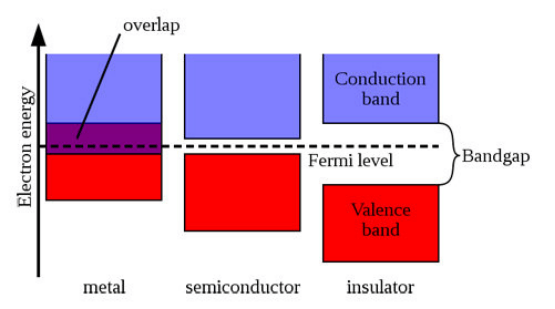
固体中的电子能级不再是离散的,而是形成连续的能带。价带(Valence Band) 是被电子占据的最高能带,导带(Conduction Band) 是空的或部分占据的最低能带。带隙(Band Gap, Eg) 是价带顶(VBM)和导带底(CBM)之间的能量差。
费米能级是温度为0 K时电子占据的最高能级,在能带图中通常标记为横虚线。它是判断材料导电性的关键参考。
下图展示了金属、半导体和绝缘体的典型能带示意图,清晰区分了带隙和费米能级的位置。

此图中,从左到右分别为金属(带重叠)、半导体(小带隙)和绝缘体(大带隙)。
二、VASP中计算能带结构的流程
VASP计算能带结构通常分两步:
自洽计算(SCF):使用密集的K点网格(如Monkhorst-Pack 8x8x8)计算电荷密度和总能量,得到稳定的波函数。输出文件包括CHGCAR、WAVECAR等。
非自洽计算(Band Structure):固定电荷密度,沿布里渊区高对称路径(k-path)采样K点,计算本征能量。
KPOINTS文件使用“Line-mode”模式,指定高对称点(如Gamma → X → L → Gamma)。
INCAR中设置:ICHARG=11(读取CHGCAR),并可能用Hybrid泛函(如HSE06)提高带隙准确性。
输出:EIGENVAL文件包含能量数据,PROCAR包含投影信息。
后处理工具:VASPKIT、pymatgen或Sumo可以自动生成k-path并绘图。常见软件如Origin或Python的matplotlib绘制能带图。
典型硅(半导体)的VASP计算能带结构示例(使用HSE泛函):

三、基于能带结构的材料分类
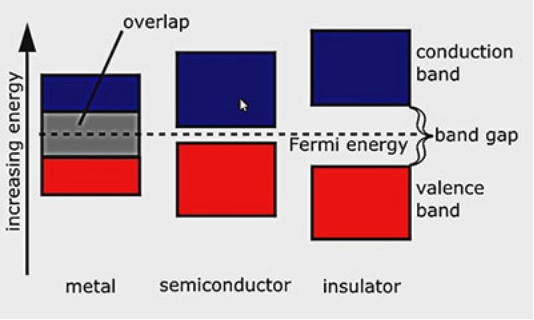
材料的电学性质主要由带隙大小和费米能级位置决定:
金属(Metal):
特征:价带和导带重叠,或费米能级穿过能带,无带隙(Eg=0)。
导电性:室温下良好导电,电子易激发。
示例:铜(Cu)、铝(Al)、金(Au)。
能带图:费米面附近有大量态。
半导体(Semiconductor):
特征:小带隙(Eg ≈ 0.1–4 eV),费米能级位于带隙中。
导电性:本征半导体导电弱,通过掺杂或温度升高可显著提高。
子类:直接带隙(VBM和CBM在同一k点,如GaAs)和间接带隙(如Si)。
示例:硅(Si, Eg≈1.1 eV)、砷化镓(GaAs, Eg≈1.4 eV)。
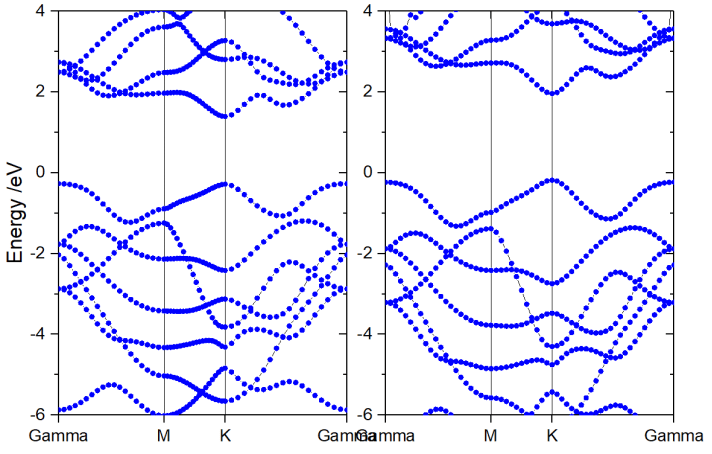
另一个硅的HSE能带图,清晰显示带隙。
绝缘体(Insulator):
特征:大带隙(Eg > 4–5 eV),费米能级在带隙中。
导电性:几乎不导电。
示例:金刚石(Diamond, Eg≈5.5 eV)、氧化铝(Al2O3)。
宽带隙半导体(如GaN, Eg≈3.4 eV)有时介于半导体和绝缘体之间,用于功率器件。
此图比较了宽带隙材料的能带特征。
半金属(Semimetal):
特征:带隙为零,但价带顶和导带底仅在特定k点接触(Dirac点或Weyl点),态密度在费米面附近很低。
导电性:弱导电,高迁移率。
示例:石墨烯(Graphene)、砷化铋(Bi)。
能带图:锥形接触点。
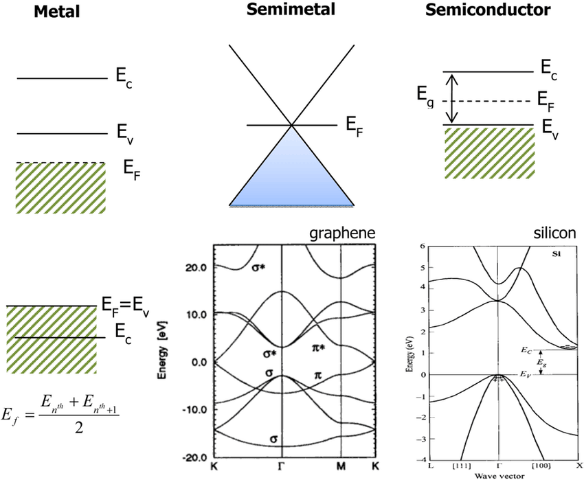
石墨烯的典型Dirac锥结构。
额外示意图:不同材料费米能级位置对比。
四、注意事项与建议
泛函选择:PBE等GGA泛函常低估带隙,推荐HSE06或GW方法提高精度。
自旋极化:磁性材料需考虑自旋向上/向下能带。
二维材料:如石墨烯,常显示线性色散关系。
收敛测试:K点密度、ENCUT对能带影响大。
通过VASP能带计算,我们能深入理解材料的电子性质,并指导新材料设计。如果你有具体材料计算疑问,欢迎讨论!


