

 474
474



芬顿(Fenton)反应作为高级氧化技术(AOPs)的核心,在环境修复、生物代谢及工业催化领域扮演着至关重要的角色。传统芬顿反应依赖铁盐与过氧化氢(H2O2)产生高活性的羟基自由基(OH),但其对 pH 值的严苛要求限制了实际应用。作为铁的同族元素,钴(Co)配合物展现出更宽的 pH 适用范围和独特的催化活性。然而,钴基类芬顿反应中活性氧的产生机制是生成游离的 OH,还是形成高价钴氧物种(Co(IV)=O)长期以来存在争议。本文章通过详细的理论计算,揭示了配体环境如何决定钴催化剂的行为模式,为新型高效催化剂的设计提供了理论支点。
研究背景
经典观点认为:
Co²⁺ 水合离子由于 Co³⁺/Co²⁺ 的氧化还原电位过高,难以像 Fe²⁺ 那样高效发生 Fenton 反应。
但实验却发现一个现象:
加入 NTA、EDTA、GSH 等配体后
Co(II)/H₂O₂ 体系可以生成 不同类型的 ROS
NTA / EDTA → 以 OOH / O₂•⁻ 为主
GSH → 以 OH 为主
👉 关键问题:
为什么不同配体会选择性地产生不同的ROS?
EDTA 体系中,为什么只有 10–15% H₂O₂ 转化为 OH?
是否存在一条非传统 Fenton-like 的主导反应路径?
计算方法
DFT 计算采用 TPSSh泛函与 def2-TZVP 全原子基组,并结合 SMD 隐式溶剂模型描述水相环境。所有结构均经几何优化与频率分析确认,并在 298 K 条件下进行自由能校正。选择 TPSSh 泛函的原因在于其对过渡金属开壳层体系具有较好的适用性,能够更可靠地描述 Co(II)/Co(III) 氧化还原过程、O–O 键断裂以及潜在的两态反应行为。
在自旋态处理方面,计算系统考察了 Co(II) 的高自旋态(S = 3/2)及 Co(III) 的不同自旋可能性,并在反应路径搜索中同时允许同自旋演化与自旋翻转。结果表明,Co(II) 向 Co(III) 转化过程中自旋态变化在能量上是可行的,其对应能垒并未构成主要动力学限制。
主要内容
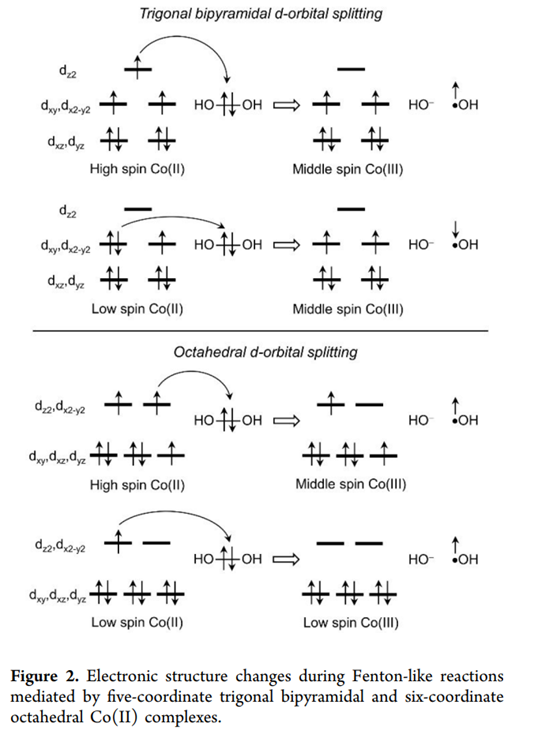
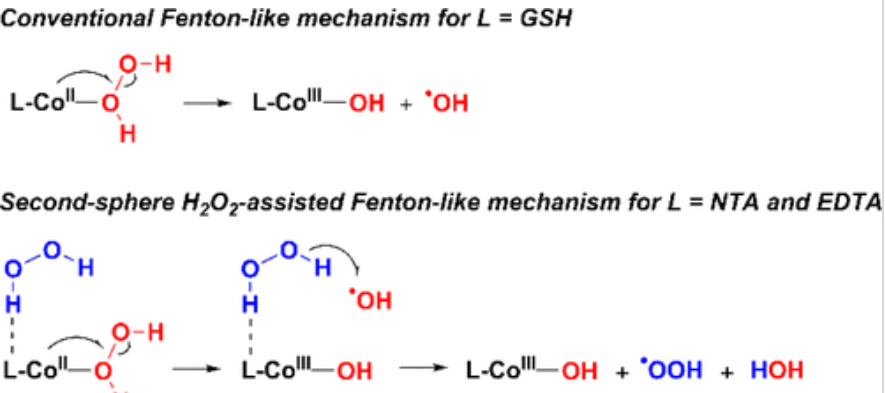
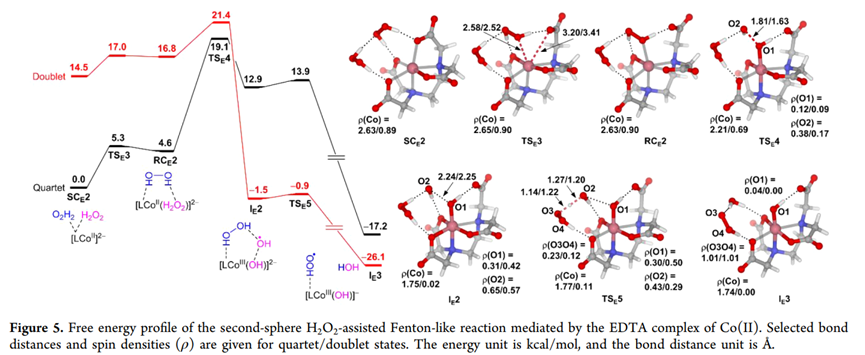
作者首次提出并系统论证了第二配位球H₂O₂ 辅助的 Fenton-like 反应机制,揭示了 Co(II) 体系中 ROS 选择性产生的根源。
三类配体的反应机制对比解读
1️⃣ NTA / EDTA:第二配位球H₂O₂ 主导 → •OOH
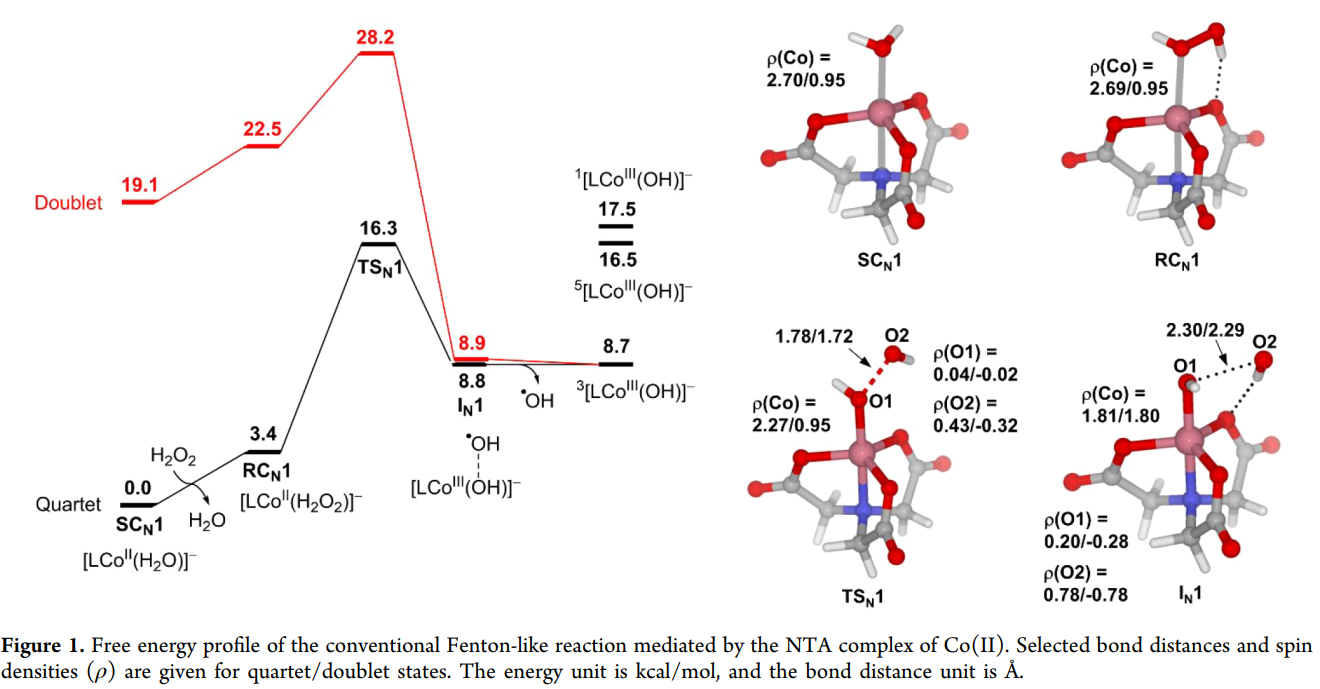
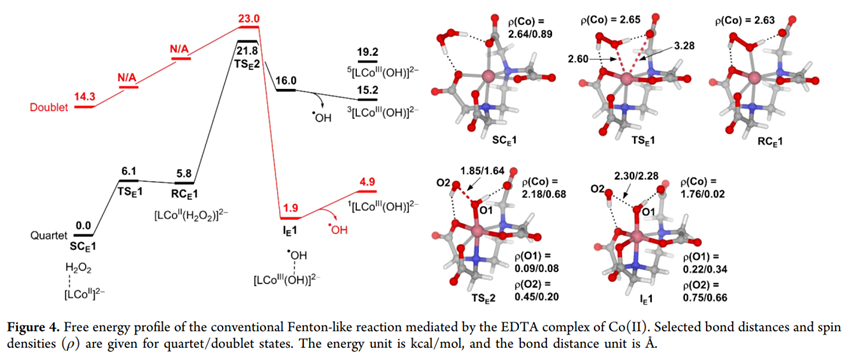
(1)传统 Fenton-like 路径:不占主导
H₂O₂ 直接配位 Co(II)
O–O 均裂 → Co(III)–OH + •OH
❌热力学不利
这解释了:
👉 为什么实验中确实检测到 •OH,但产率很低
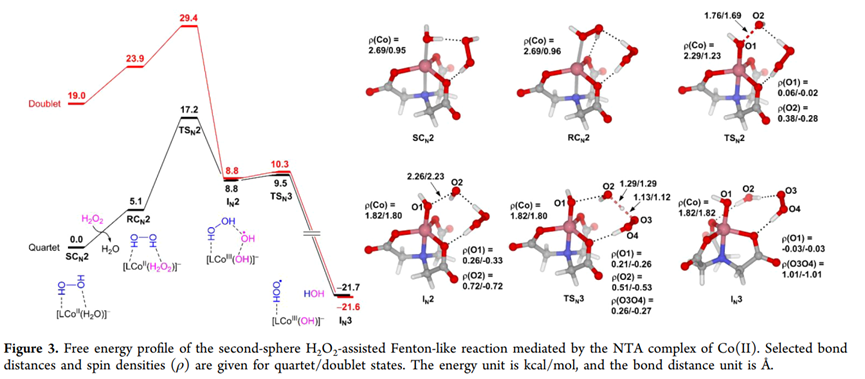
(2)关键发现:第二配位球H₂O₂ 的作用
NTA / EDTA 含多个 羧酸盐基团,可以:
通过氢键 捕获额外的 H₂O₂
在金属中心附近形成 H₂O₂ 富集区
一旦产生瞬态 •OH,会立刻发生:
OH + H₂O₂ → H₂O + •OOH(HAT 反应)
✔ 强烈放热,提供反应热力学驱动力
✔ 阻止 •OH 回配位
✔ 解释 •OOH / O₂•⁻ 为主ROS 的实验现象
2️⃣ EDTA 体系的10–15% •OH?
计算结果明确指出:
传统 Fenton-like 路径确实存在
但并非主路径
主反应路径仍然是:
👉第二配位球H₂O₂ → •OOH
这完美解释了实验中:
OH 可检测,但只占 H₂O₂ 消耗的一小部分
3️⃣ GSH:硫醇配位直接降低门槛→ •OH
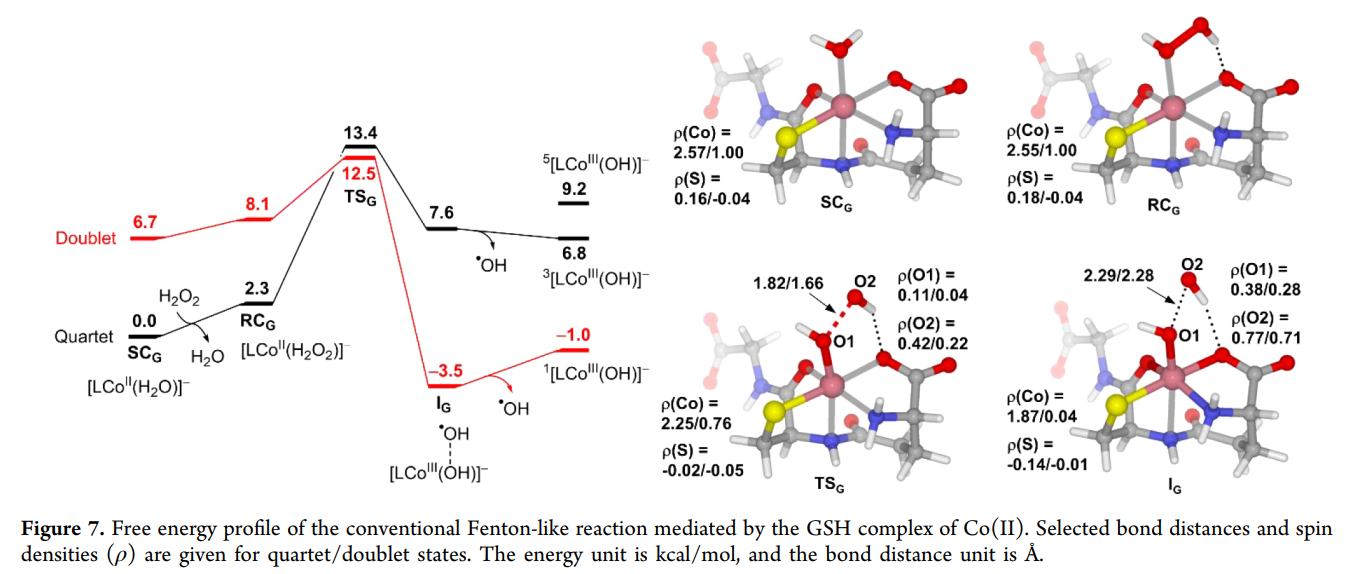
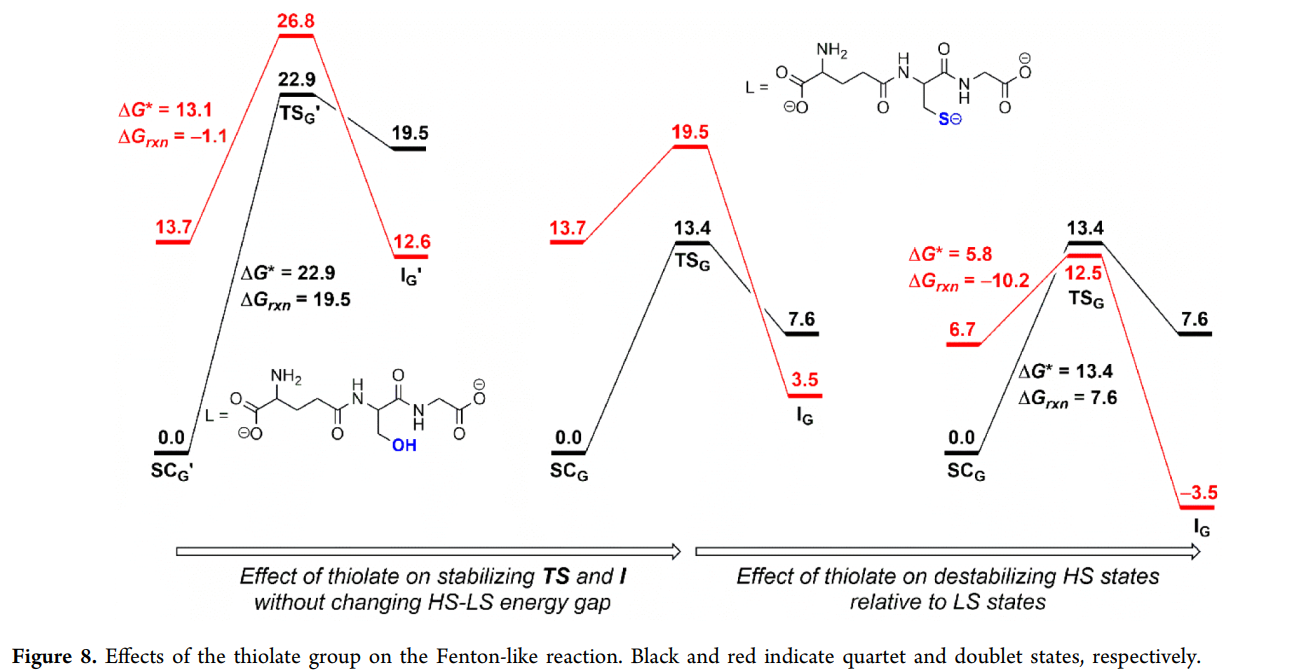
GSH 的关键不在第二配位,而在:
S⁻直接配位 Co(II)
DFT 对比计算表明:
仅有 S⁻ 能显著降低:
O–O 断裂能垒
反应自由能
若将 S⁻ 替换为 –OH → 反应立刻变得不利
促进能力排序(强 → 弱):
S⁻ > –NH₂ > –CONH– > –COO⁻ > C=O
📌 结论:
GSH 体系是经典的 Co(II)-Fenton-like 反应,•OH 为主 ROS
明确区分了:第一配位调控与第二配位调控
合理引入:
两态反应
自旋翻转在 Co 体系中是允许且可行的
理论结果与EPR、自旋捕获、光谱实验高度一致
总结
该工作通过系统 DFT 计算,首次区分并量化了 Co(II)-Fenton-like 体系中第一与第二配位对反应路径的本质影响。计算结果表明,传统 O–O 均裂路径在 Co(II) 体系中并非主导,而第二配位球 H₂O₂ 可通过后续放热的 HAT 过程重塑整体自由能面,从而解释 •OOH 为主 ROS 的实验现象。相比之下,thiolate 配位通过直接调控金属中心电子结构,使经典 •OH 生成路径重新变得可行。



