

 972
972

一、研究背景
氨合成是可持续能源体系的关键环节,传统哈伯 - 博施法依赖高温高压条件,消耗全球近 2% 的能源并排放 3% 的 CO₂,亟需开发温和条件下的绿色合成技术。
电催化氮还原反应(NRR)是低能耗、零排放分布式氨合成的理想途径,但面临 N≡N 键(键能 941 kJ/mol)活化动力学缓慢、水溶液中析氢反应(HER)竞争剧烈的问题,导致催化活性与选择性偏低。
单原子分子催化剂(SAMCs)凭借原子级精准调控配位环境的优势,成为提升 NRR 选择性的核心研究方向。其中,碳硼龙(carbolong)家族分子(如含 15 个碳原子的 carborin)具有独特共轭骨架与金属中心整合结构,为协同催化提供了多功能平台。
传统密度泛函理论(DFT)模拟中,计算氢电极(CHE)方法假设每步电子转移固定为 1e⁻,忽略了实际电催化过程中电位驱动的电荷态变化,导致中间体自由能计算不准确,甚至误判速率决定步骤(PDS);且缺乏高效方法整合电压效应、溶剂效应与催化剂 - 载体相互作用,难以系统筛选高性能 NRR 催化剂。
二、研究亮点
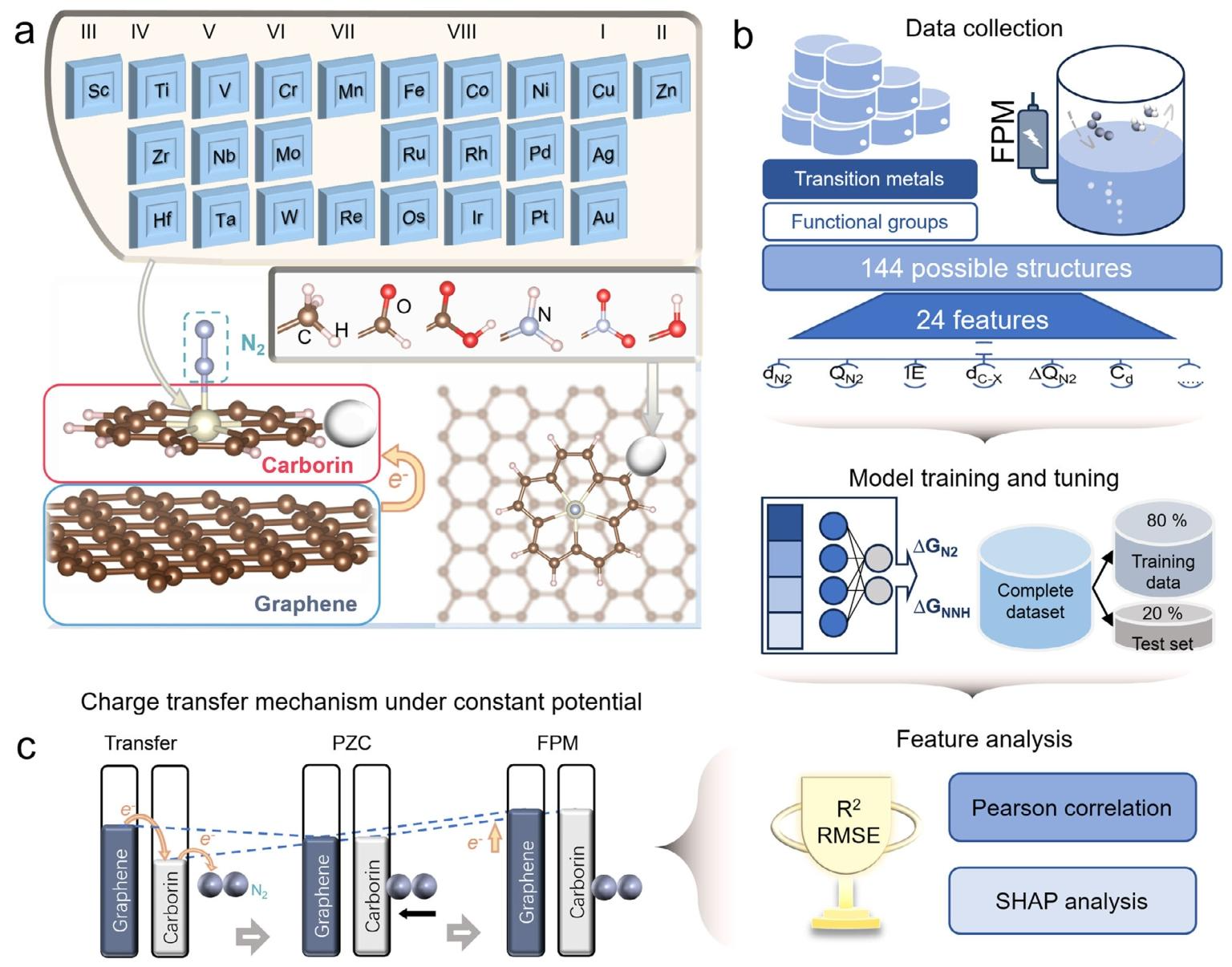
创新集成研究框架:首次将机器学习(ML)与正则系综恒电位法(FPM)相结合,构建 DFT-ML 高通量筛选体系,同时考虑电位效应、溶剂效应与界面电子耦合,实现 NRR 催化剂的精准筛选与机制解析。
高效筛选最优催化剂:设计并筛选 144 种官能团修饰的 carborin / 石墨烯负载单原子催化剂(涵盖 25 种过渡金属与 6 种官能团),发现 Cr@NO₂-carborin / 石墨烯与 Cr@CHO-carborin / 石墨烯性能最优,限制电位分别低至 - 0.220 V(*N₂→*N₂H 步骤)与 - 0.245 V(*NH→*NH₂步骤),NRR 对 HER 的法拉第效率高达 99%。
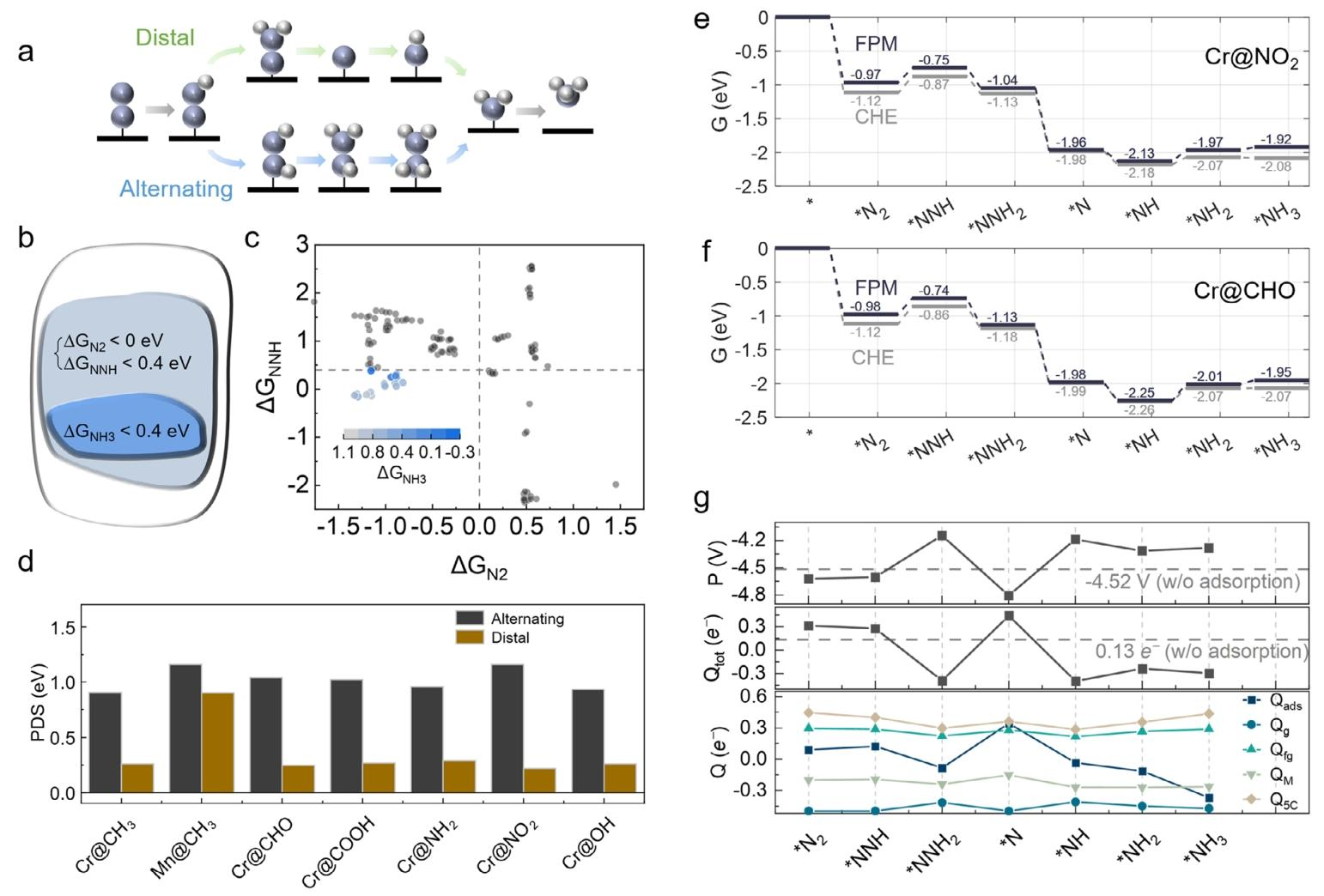
纠正传统方法缺陷:对比 FPM 与传统 CHE 方法,证实 CHE 因忽略中间体电荷态变化,会导致自由能计算偏差与 PDS 误判(如将 Cr@CHO-carborin 的 PDS 误判为 * N₂→NNH,实际为NH→*NH₂),而 FPM 可精准描述恒电位下的电荷转移动态过程。
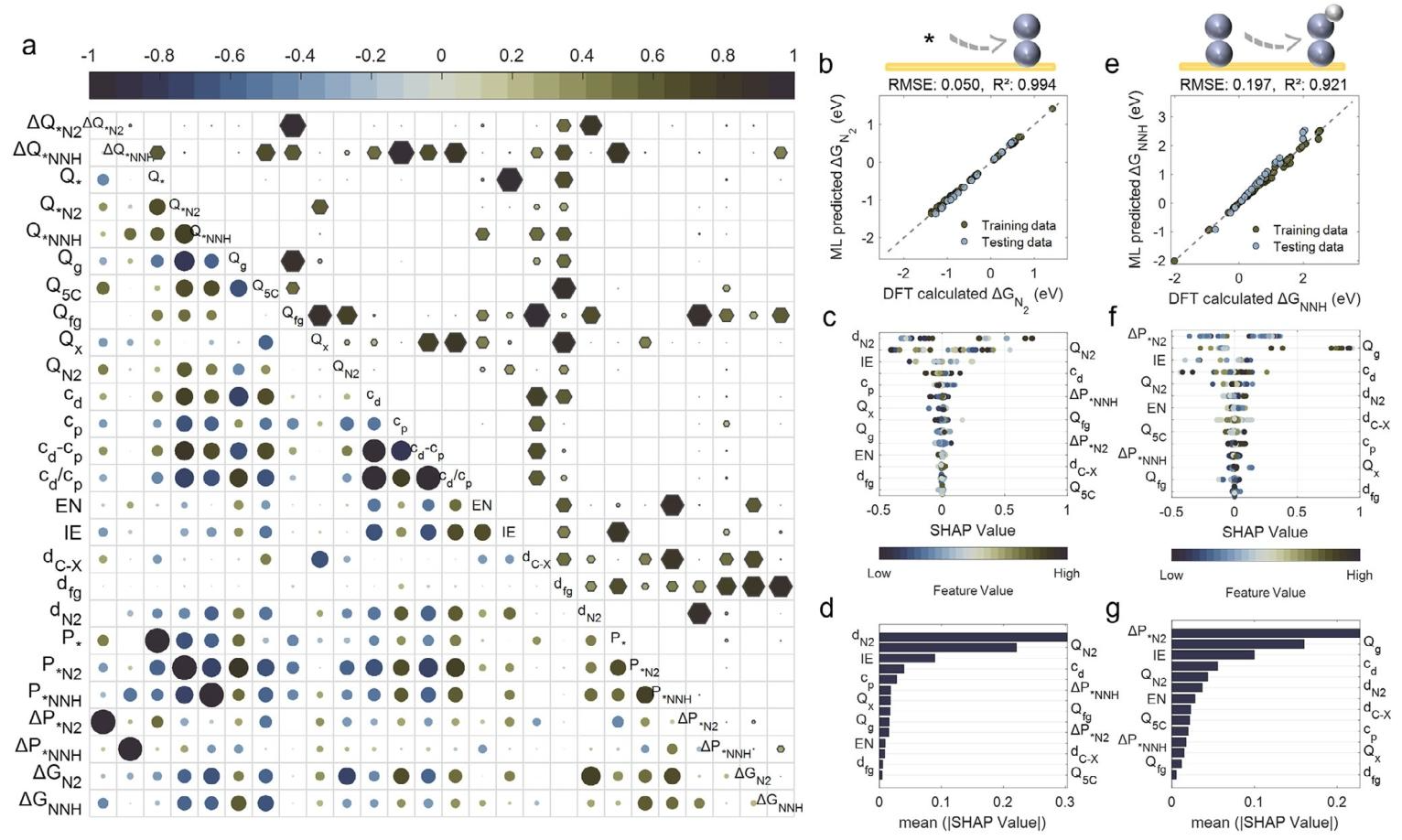
发现关键动态描述符:通过 Pearson 相关性分析与 SHAP 解释性模型,揭示中间体吸附诱导的零电荷电位(PZC)偏移是调控电荷转移模式与 N₂活化的核心电压响应描述符,突破了传统静态电子描述符的局限。
验证催化剂稳定性:通过 300 K 下 5000 fs 的从头算分子动力学(AIMD)模拟,证实最优催化剂具有优异的热力学稳定性,且 NH₃脱附过程在热力学上可行。
三、核心机制
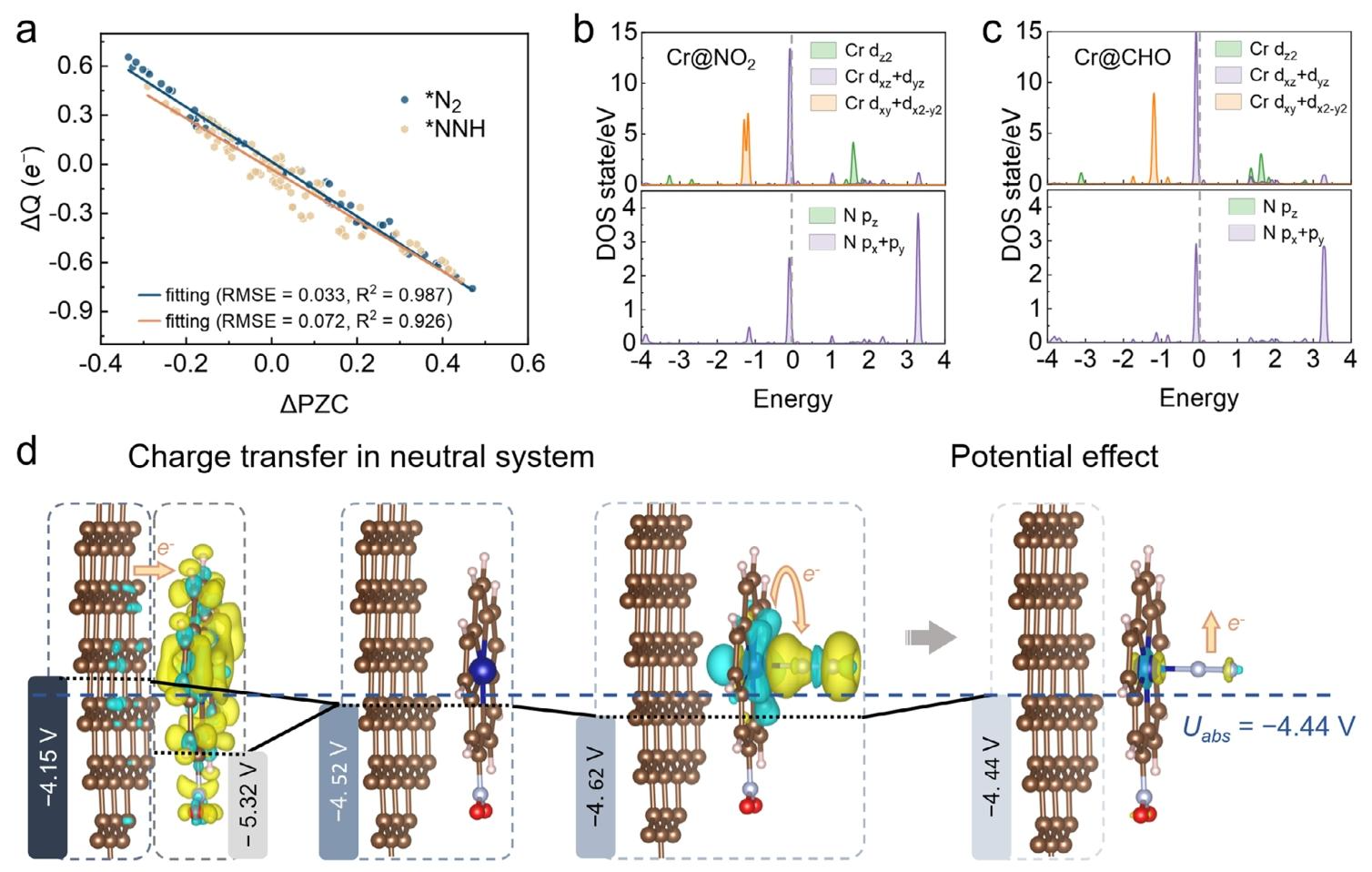
恒电位下的电荷转移动态调控:FPM 方法明确,恒电位条件下中间体(*N₂、*NNH 等)的吸附会诱导 PZC 偏移,进而改变体系电荷态(从 n₁e⁻变为 n₂e⁻),导致净电荷转移量(n₂-n₁-1)e⁻与 CHE 假设的固定 - 1e⁻存在显著差异,直接影响中间体自由能与反应动力学。
PZC 偏移主导催化活性:PZC 变化与体系电荷变化呈强线性相关(R² 分别为 0.987 和 0.926),Cr@NO₂-carborin 吸附 N₂后 PZC 从 - 4.52 V 降至 - 4.62 V,引发负电荷积累(从 0.132 e⁻增至 0.316 e⁻),适度削弱 N₂吸附强度,同时促进电子从 Cr 的 d 轨道向 N₂的 p 轨道转移,弱化 N≡N 键。
活性中心电子结构优化:最优催化剂中 Cr 的 d 带中心处于火山曲线底部,对 * NNH 中间体具有最优结合强度,平衡了中间体活化与产物脱附;Cr 的 d 轨道与 N 的 p 轨道在费米能级附近显著杂化,进一步强化 N₂活化。
反应路径选择性机制:NRR 遵循能量更有利的远端路径,交替路径因 * NHNH 中间体形成需更高 PDS 而动力学不利;石墨烯载体的高导电性促进层间电荷重分布,显著影响活性位点的局部电子环境,提升催化动力学。
四、总结与意义
本研究通过机器学习与恒电位 DFT 模拟的创新性集成,系统探究了官能团修饰 carborin / 石墨烯负载单原子催化剂的 NRR 性能,筛选出 Cr@NO₂-carborin / 石墨烯与 Cr@CHO-carborin / 石墨烯两种高性能催化剂。研究证实,恒电位下中间体吸附诱导的 PZC 偏移是调控电荷转移与 N₂活化的核心描述符,而传统 CHE 方法因忽略电荷态动态变化存在显著局限性。FPM 与 ML 的结合不仅实现了催化剂的高效筛选,更揭示了 NRR 的动态电荷转移机制,为多电子转移电催化反应的机制解析提供了通用框架。
论文DOI: 10.1002/advs.202524356


