

 1252
1252

研究背景
固有手性分子(尤其是中环化合物)因具有刚性三维结构,在不对称催化中展现出潜在应用价值却长期被忽视。含氮八元环广泛存在于天然产物、生物活性分子和功能材料中,当这类分子缺乏手性碳等传统手性单元,但环上富$sp^2$碳时,会形成稳定的鞍形构象从而表现出固有手性。目前这类固有手性八元氮杂环的对映选择性合成仍面临挑战,虽已有不对称成环、动力学拆分(KR)/动态动力学拆分(DKR)等方法,但针对10-取代三苯并氮杂䓬类固有手性化合物的高效合成方法仍待开发。
不对称氢化是构建碳基手性中心的重要手段,本文将其应用于无新立体中心生成的简单烯烃,借助手性催化剂对原有手性单元的识别,可实现外消旋体的动力学拆分,而亚氨基氮的氢键位点为手性磷酸(CPA)催化的不对称转移氢化提供了可行性。
核心问题
如何开发基于CPA催化的不对称转移氢化策略,实现固有手性10-乙烯基三苯并氮杂䓬的高效动力学拆分?
明确该动力学拆分反应的最优条件、底物适用范围及官能团耐受性。
阐释氢化产物在酸性条件下易消旋、而未反应底物光学稳定的原因及具体机理。
研究方法和主要内容
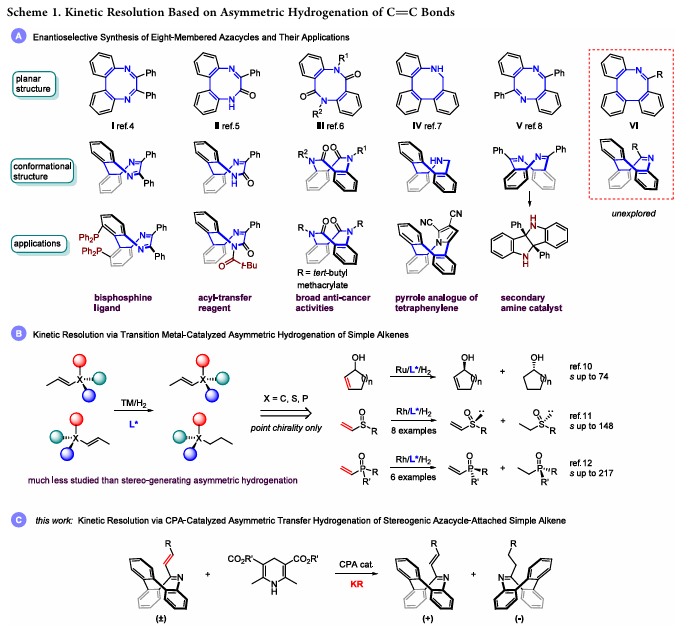
反应条件优化与核心结果
以外消旋10-(E-2-苯乙烯基)三苯并氮杂䓬(1a)为模型底物,汉斯酯(HE)为氢源,系统筛选CPA催化剂、氢源、溶剂等反应条件,核心优化结果如下:
- 最优催化剂:含9-蒽基的BINOL衍生CPA(C3)对映选择性最优,其他CPA催化剂效果显著降低。
- 最优氢源:汉斯甲酯(HE2)为氢源时拆分效果最佳,叔丁酯(HE4)因空间位阻完全无氢化活性。
- 最优溶剂:甲苯为溶剂时,未反应底物(+)-1a的对映体过量值(ee)达99%(收率47%),氢化产物(-)-2a收率52%(ee77%)。
- 关键添加剂:5Å分子筛是抑制氢化产物消旋的关键,移除后氢化产物完全消旋,而底物光学纯度基本不受影响。
最优反应条件:(±)-1(0.1 mmol)、HE2(0.6当量)、C3(10 mol%)、5Å分子筛(25 mg),甲苯为溶剂,25℃反应24 h。
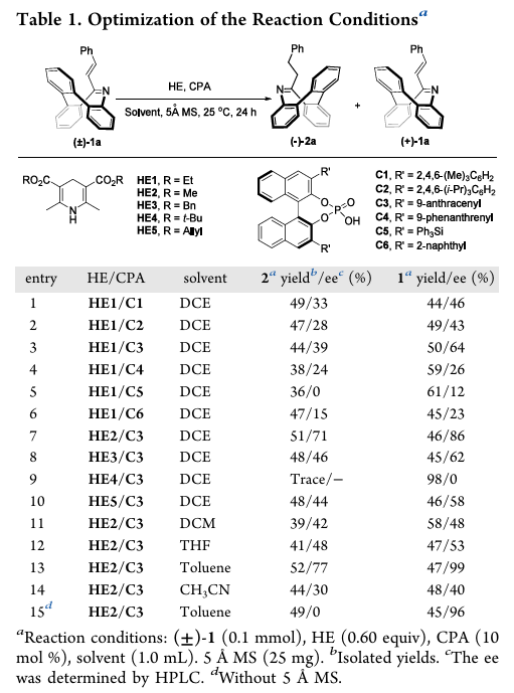
底物范围与官能团耐受性
在最优条件下,考察了10-位烯烃取代基、氮杂环骨架修饰的底物兼容性,均表现出良好的反应效率和高对映选择性:
- 苯乙烯基取代底物:苯环对位含给电子基(-Me、-OMe)、吸电子基(卤素、-CF₃、-NO₂)的底物均能耐受,未反应底物ee达90%以上;间位/邻位取代、萘环替代苯环的底物对反应无明显影响,ee保持90~97%;吡啶环取代因与CPA发生竞争性氢键作用,完全抑制氢化反应。
- 烷基取代烯烃底物:不同链长烷基、环丙基、酯基取代的乙烯基底物均能实现对映选择性氢化,动力学拆分效果良好;存在分子内氢键的顺式烯醇式底物完全无反应活性,证实氢键位点对CPA催化的重要性。
- 氮杂环骨架修饰底物:环上C12、C13位的各类取代底物均适用于该反应,未反应底物ee达90%~>99%,展现出优异的骨架兼容性。
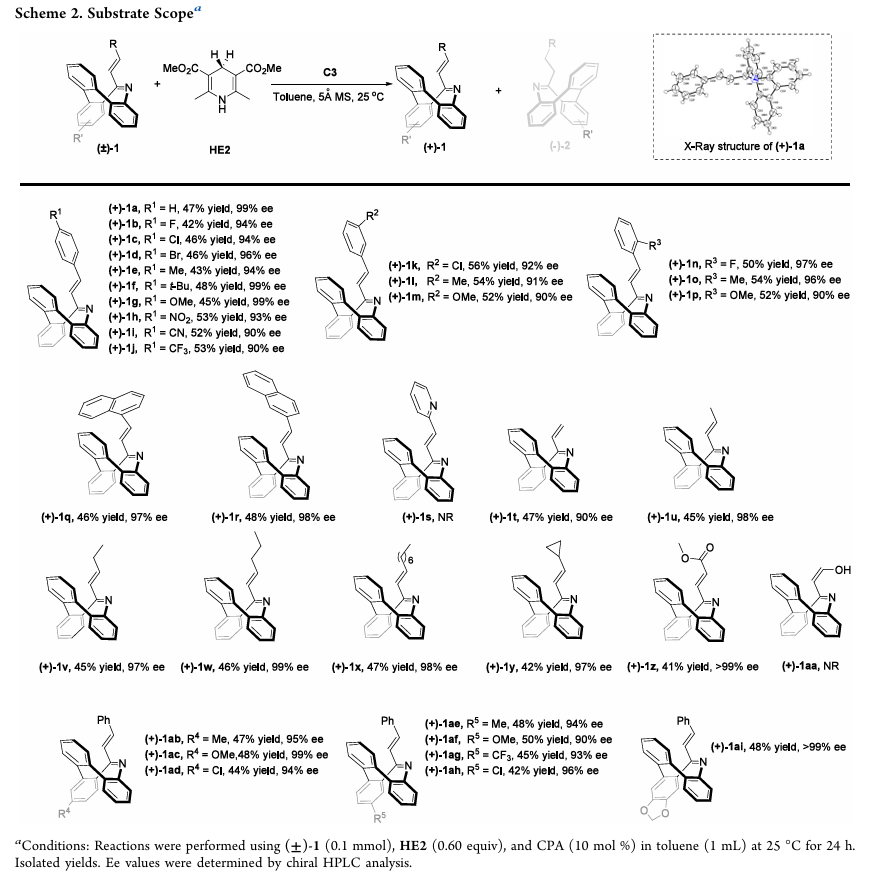
反应机理与消旋原因探究
(1)动力学拆分反应机理
CPA通过氢键同时活化底物中的α,β-不饱和亚胺基和汉斯酯,形成过渡态后对映选择性识别外消旋底物的两个立体异构体,发生立体可控的1,4-共轭加成生成烯胺中间体,经互变异构得到氢化产物(-)-2a;而另一种立体异构体因与催化剂匹配性差未发生反应,最终实现外消旋氮杂䓬的动力学拆分。
(2)氢化产物消旋机制
- 实验验证:氢化产物(-)-2a在甲苯中热稳定性优异,消旋能垒ΔG达32.4 kcal/mol(110℃下半衰期38.7 h);但在水(5.0当量)和磷酸二苯酯(10 mol%)存在下,20℃时半衰期仅16.5 min(ΔG=21.8 kcal/mol),快速消旋;未反应底物(+)-1a在相同酸性条件下因水分子更易对其双键发生1,4-加成,反而表现出光学稳定性。
- DFT计算:酸性条件下,水分子对氢化产物亚胺基的亲核加成是速率控制步骤,能垒为22.3 kcal/mol,与实验值(21.8 kcal/mol)高度吻合;亲核加成生成的水合中间体易发生开环,进而经关环实现消旋,揭示了氢化产物酸敏性的本质。
实用性验证与产物转化
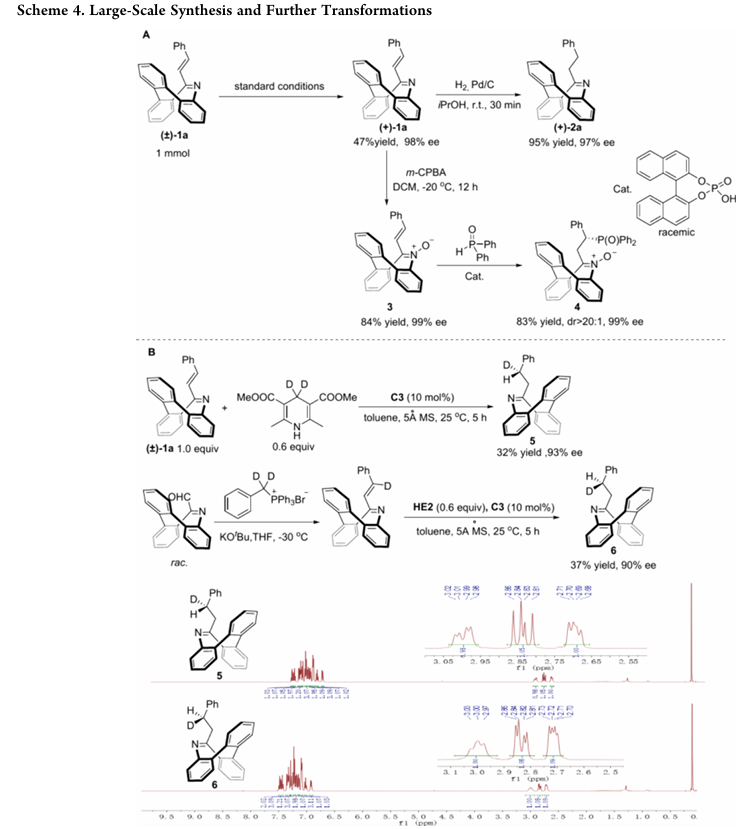
- 放大反应:将反应规模放大至1 mmol,仍能高效实现动力学拆分,(+)-1a收率47%、ee 98%,展现出良好的工业应用潜力。
- 产物衍生化:
无酸条件下,(+)-1a经Pd/C/H2氢化可高收率(95%)、高对映选择性(ee97%)得到(+)-2a;
(+)-1a经m-CPBA氧化生成N-氧化物3(收率84%,ee99%),其与二苯基氧化膦的迈克尔加成能非对映选择性生成产物4(收率83%,无对映体流失);
利用氘代汉斯酯和氘代底物,可成功合成同位素非对映体5和6,且能通过NMR光谱明确区分。
结论
开发了一种基于CPA催化的不对称转移氢化策略,实现了固有手性10-取代三苯并氮杂䓬的高效动力学拆分。该方法无需生成新的立体中心,通过CPA对鞍形氮杂环的精准识别,实现了烯烃侧链的对映选择性氢化,反应条件温和、官能团耐受性广、收率和对映选择性优异(未反应底物ee最高达99%)。明确了氢化产物在酸性条件下因亚胺基的酸促水合-开环-关环过程易消旋,而未反应乙烯基底物在相同条件下光学稳定的构效关系;通过无酸氢化可高对映选择性获得氢化产物,DFT计算和实验研究阐明了氢化产物的消旋机理。
ACS Catalysis. 中科院广州生物医药与键康所罗爽、朱强


