

 3686
3686
晶体轨道哈密顿布居(Crystal Orbital Hamilton Population,简称COHP)是一种广泛应用于固体化学和材料科学中的量子化学分析方法。它能够从密度泛函理论(DFT)等电子结构计算结果中,直接提取原子间成键、反成键和非成键相互作用的信息,并以能量分辨的方式可视化呈现。COHP的核心思想是将晶体轨道的哈密顿矩阵元素(H_μν)加权到态密度上,从而将带结构能量按化学键类型进行分区。1993年,Richard Dronskowski等人首次提出这一方法,随后通过LOBSTER等软件实现与平面波基组(如VASP)的兼容(pCOHP),使其成为分析间金属化合物、相变材料和磁性材料等复杂体系的强大工具。
(https://www.mdpi.com/2073-4352/8/5/225)
COHP与COOP的异同:从重叠到哈密顿的演进
COHP与著名的晶体轨道重叠布居在概念上高度相似,都是将晶体轨道按原子对进行投影,以揭示键合性质。但二者关键区别在于加权因子:COOP使用重叠矩阵元素(S_μν),成键表现为正值(正重叠);而COHP使用哈密顿矩阵元素(H_μν),成键对应负值(能量降低)。因此,实际绘图时常用–COHP,使成键峰向右(正值)、反成键向左(负值),便于与COOP曲线直观对比。COHP的优势在于它直接与带结构能量相关,更适合DFT计算,且对基组依赖更小,尤其在平面波方法中通过投影重建哈密顿矩阵实现高精度分析。
COHP的理论基础与计算实现
在 Bloch 定理框架下,晶体波函数由原子轨道线性组合而成,COHP通过以下公式定义(针对原子对A-B):
COHP_{A-B}(E) = ∑_μ∈A ∑_ν∈B H_μν P_μν(E)
其中P_μν(E)为密度矩阵,H_μν为哈密顿矩阵元。对能量的积分(ICOHP)可定量给出特定键的键强度(单位eV),正如积分DOS给出总电子数一样。实际计算中,先用VASP、Quantum ESPRESSO等软件完成自洽DFT计算,再通过LOBSTER程序投影到局域辅助基组,自动生成各原子对的COHP曲线、集成值(ICOHP)及百分比贡献。该方法已广泛用于高通量键合分析。
如何解读COHP曲线:成键 vs 反成键
在–COHP图中,费米能级(E_F)以下的正峰表示成键贡献(稳定结构),负峰表示反成键(不稳定);E_F处的反成键峰往往暗示结构不稳定或相变驱动因素。非成键区域接近零。集成值ICOHP可直接比较键强度,例如在极性间金属化合物中,异原子键(如T-R)通常主导成键,而同原子键(如R-R)贡献较小。通过与DOS对比,还能揭示磁性、电子计数规则(e/a比)等对键合的影响。
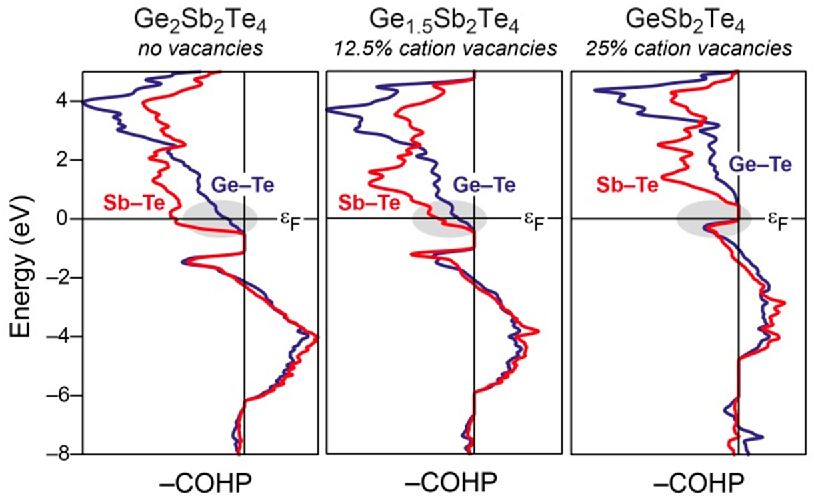
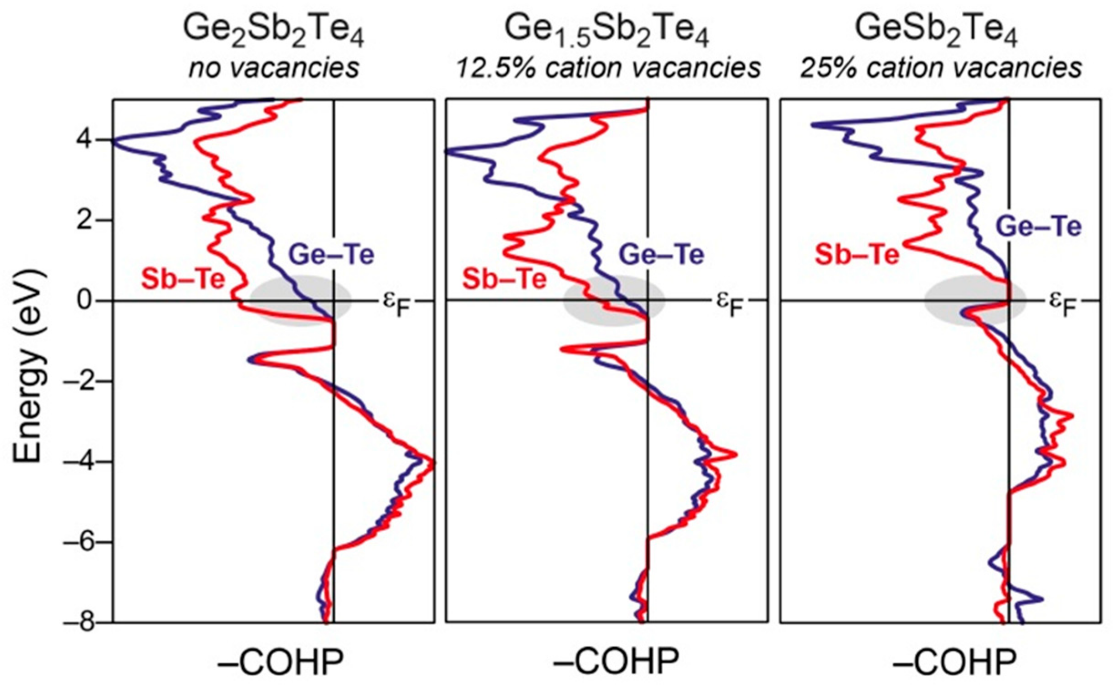
实际应用示例1:相变材料Ge-Sb-Te体系中的键合调控
在相变存储材料Ge₂Sb₂Te₄、Ge₁.₅Sb₂Te₄和GeSb₂Te₄中,COHP分析揭示了阳离子空位对Ge-Te和Sb-Te键的影响。随着空位增加,费米能级处的反成键相互作用减弱,结构稳定性提升。这直接解释了为何富锗相更易发生非晶-晶态转变。–COHP曲线中蓝色(Ge-Te)和红色(Sb-Te)峰清晰显示了空位诱导的键合优化,是材料设计中典型的COHP应用。
(https://www.mdpi.com/2073-4352/8/5/225)
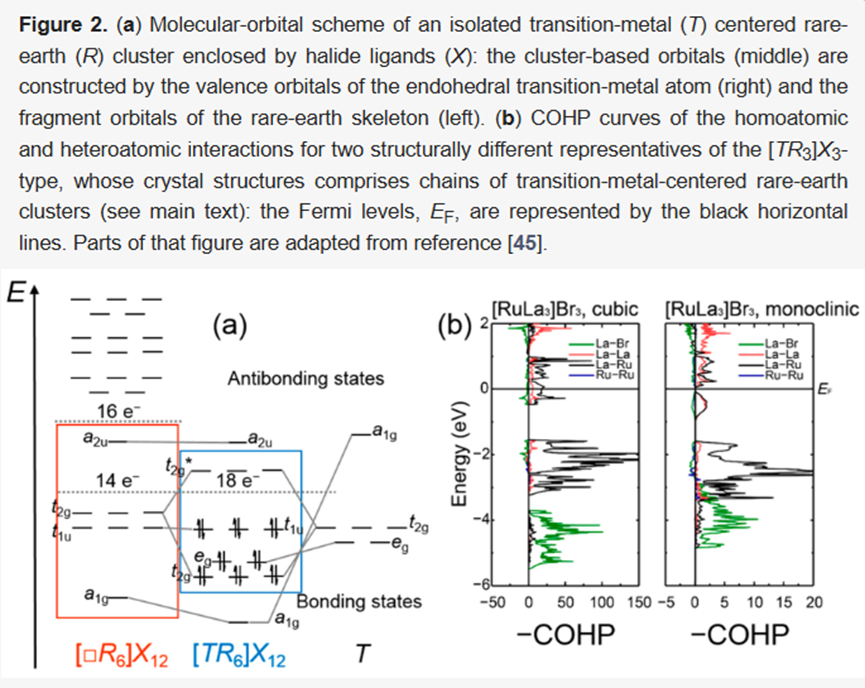
实际应用示例2:稀土过渡金属卤化物簇合物中的成键分析
在[TR₆]簇(T=过渡金属,R=稀土)及其聚合链结构中,COHP曲线显示T-R异原子相互作用主导成键,R-R键贡献次之,而卤素配体主要起离子作用。通过比较立方型与单斜型[TR₃]X₃结构,COHP还解释了费米能级位于DOS峰或隙的电子稳定性差异,为金属化合物的价电子计数提供了精确补充。
(https://www.mdpi.com/2073-4352/8/5/225)
如今,在磁性材料(如FeN)、铁电体和催化剂中,COHP帮助揭示自旋极化如何消除EF处反成键态,从而稳定铁磁序。相比传统Bader电荷或Mulliken分析,COHP提供能量直接、轨道直观的键强度量化,成为不可或缺的键合显微镜。


