

 640
640
比旋光度(specific optical rotation,[α])这一性质在手性分子鉴定、药物研发以及天然产物绝对构型解析中具有重要意义。比旋光度实验可以测量,而理论计算则可以提前预测构型(R/S),大大降低实验成本。本文将以 Gaussian(G09/G16 通用)为核心,系统讲解:基本原理、计算流程 、注意事项等。
一、什么是比旋光度?
手性分子能够使平面偏振光发生旋转,这种现象称为光学活性(optical activity)。
比旋光度定义为:
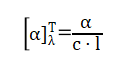
其中:
α:观测旋光度(单位:°)
c:浓度(g/mL)
l:光程(dm)
λ:光的波长(常用 589 nm,即钠 D 线)
T:温度(通常为 25℃)
补充理解
比旋光度随波长变化,这种现象称为旋光色散(ORD)
不同构型(R/S)对应符号相反的旋光度
为什么要计算?
实验测量比旋光度存在一定局限:
需要纯样品
需要专用旋光仪
对复杂或不稳定分子不友好
而理论计算可以:
直接从分子结构预测
辅助判断绝对构型
处理难以分离的体系
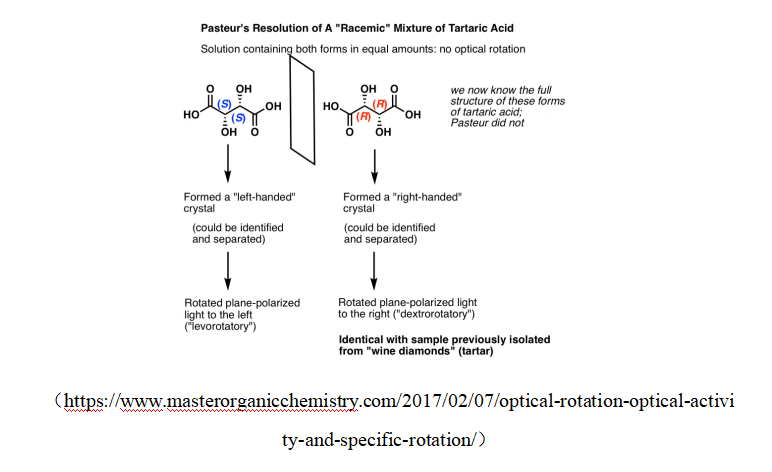
(https://www.masterorganicchemistry.com/2017/02/07/optical-rotation-optical-activity-and-specific-rotation/)
二、Gaussian 计算比旋光度的理论基础
Gaussian 中比旋光度的计算基于 Rosenfeld 方程,本质涉及:
电偶极矩
磁偶极矩
极化率张量
在实际计算中,Gaussian 通过:
TD-DFT来高效实现光学旋转性质的计算。

(https://www.masterorganicchemistry.com/2017/02/07/optical-rotation-optical-activity-and-specific-rotation/)
三、Gaussian 计算流程
几何优化 + 频率分析
目的:
获得稳定构型
确认无虚频(真正极小值)
比旋光度计算
核心关键词:
Polar=OptRot
CPHF=RdFreq
说明:
Polar=OptRot:计算光学旋转
CPHF=RdFreq:指定计算波长(频率依赖)
结果提取
在输出文件(.log)中搜索:
[Alpha]
或:
Optical Rotation
即可得到比旋光度(单位):
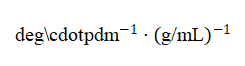
此外可通过 GaussView:
👉 Results → ORD
绘制旋光色散曲线
四、推荐计算参数
泛函选择
APFD(推荐)
B3LYP(常用)
CAM-B3LYP(适合激发态)
基组选择
几何优化:
6-311+G(2d,p)
光学计算:
6-311++G(2d,2p)
或 aug-cc-pVDZ(更推荐)
👉 关键点:必须包含弥散函数(+ / ++)
溶剂模型
PCM
SMD
👉 建议加入溶剂效应以接近实验条件
波长设置
常用多波长计算:
355 nm
589 nm
633 nm
👉 用于绘制 ORD 曲线
五、经典案例:甲基环氧乙烷(Methyloxirane)
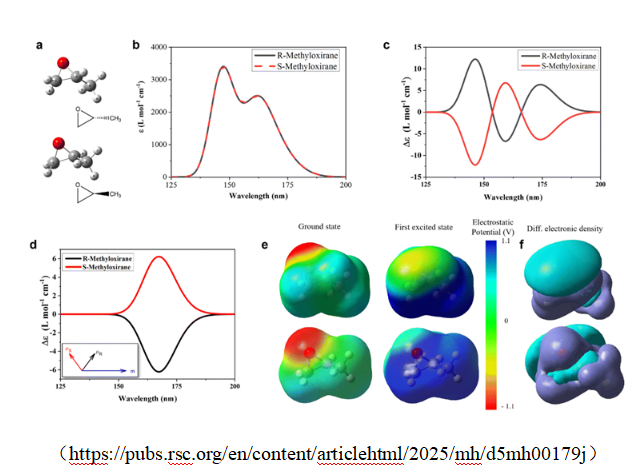
(https://pubs.rsc.org/en/content/articlehtml/2025/mh/d5mh00179j)
几何优化输入(S 构型)
%nprocshared=8
%mem=1GB
# opt freq apfd/6-311+g(2d,p)
S-methyloxirane optimization
0 1
(分子坐标)
比旋光度计算输入
%nprocshared=8
%mem=1GB
# apfd/6-311++g(2d,2p) polar=optrot cphf=rdfreq
S-methyloxirane OR
0 1
(优化后坐标)
355nm 633nm
结果对比
| 波长 | 计算值(S) | 实验值 |
| 355 nm | +4.85 | +7.49 |
| 633 nm | -11.3 | -8.39 |
👉 关键结论:
R 构型数值符号相反
可通过“符号 + 趋势”判断绝对构型
注意事项
基组依赖性
小基组误差明显
推荐使用含弥散函数基组
构象平均
对于柔性分子:
👉 必须进行 Boltzmann 加权
否则结果可能严重偏差
溶剂效应
气相结果往往偏离实验
建议加入 PCM 或 SMD
高精度方法
CCSD 等方法精度更高
但计算成本极大
👉 实际中 TD-DFT 已足够实用
输出读取技巧
在 .log 文件中查找:
Optical Rotation
[Alpha]
扩展体系
多种环氧化物(oxirane 衍生物)
天然产物
药物分子
👉 Gaussian 均表现出良好预测能力
实际应用
手性药物构型判断
天然产物结构解析
手性纯度辅助分析
总结
Gaussian 的 Polar=OptRot 功能,使比旋光度的理论计算变得:高效、可操作 与实验高度互补 掌握这一方法可以:
👉 在实验之前预测手性分子的光学性质
👉 通过计算 + 实验双重验证绝对构型
在现代有机化学与药物研发中,这已经是一项非常重要的核心技能。



