

 325
325



多手性中心的吲哚啉/β-咔啉骨架广泛存在于天然产物和药物分子中,但其合成通常依赖多步反应逐步构建,效率较低。近年来,自串联催化因能够在单一催化剂作用下实现多个机理不同的反应步骤,避免中间体分离,被认为是提高合成效率的重要策略。然而:不对称自串联催化仍然发展有限;多反应过程之间的兼容性与选择性控制困难因此,发展新的不对称自串联催化体系具有重要意义。
研究背景
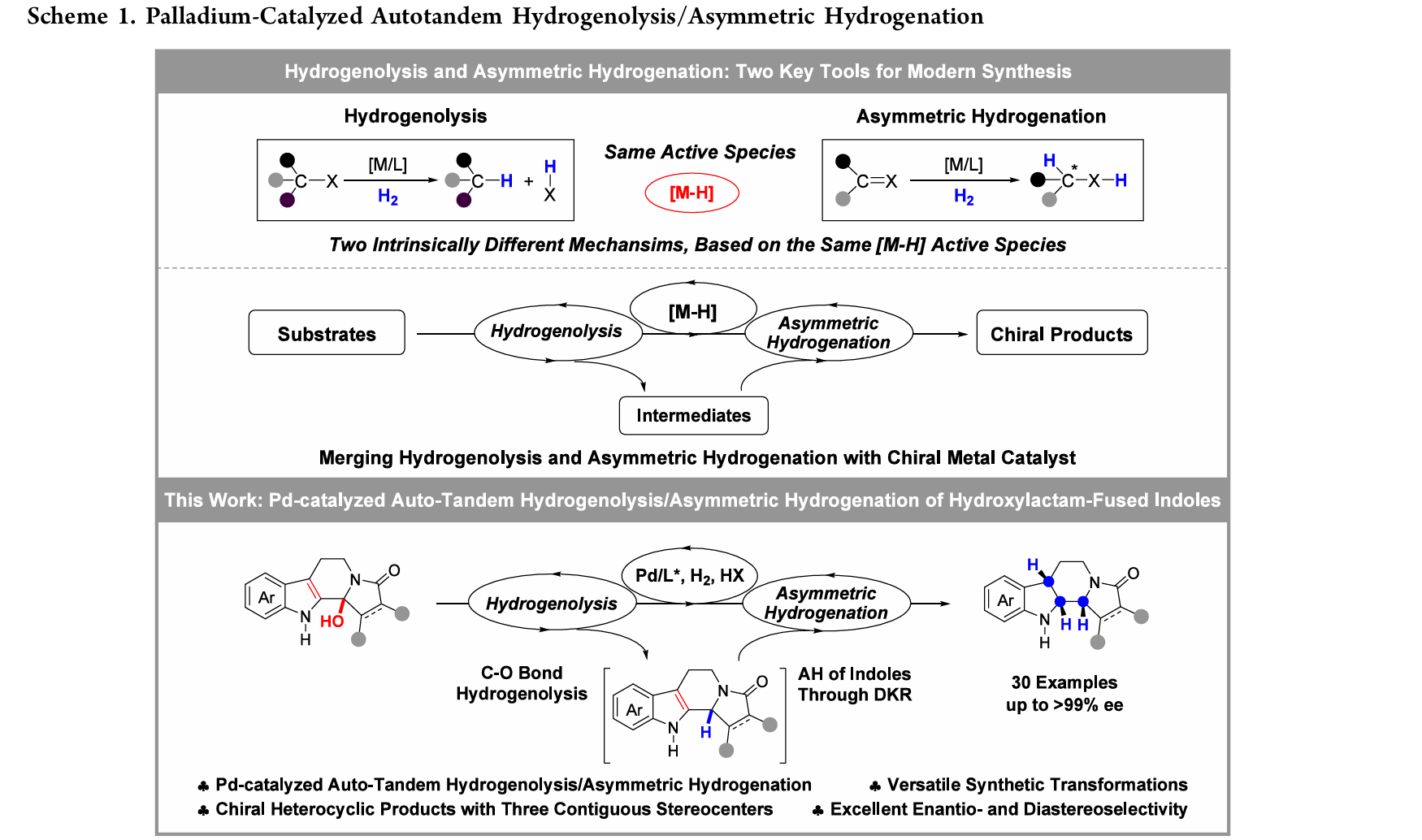
作者的核心设想是:
👉 利用同一种活性物种——Pd–H
同时实现:
C–O键氢解
吲哚的不对称氢化
并结合:
👉 动态动力学拆分(DKR)
从而在一步反应中构建多个连续手性中心。
关键挑战包括:
多步骤反应的顺序、协同及化学选择性控制
Pd–H对不同反应的兼容性
化学选择性、对映选择性和非对映选择性的统一控制
主要研究内容
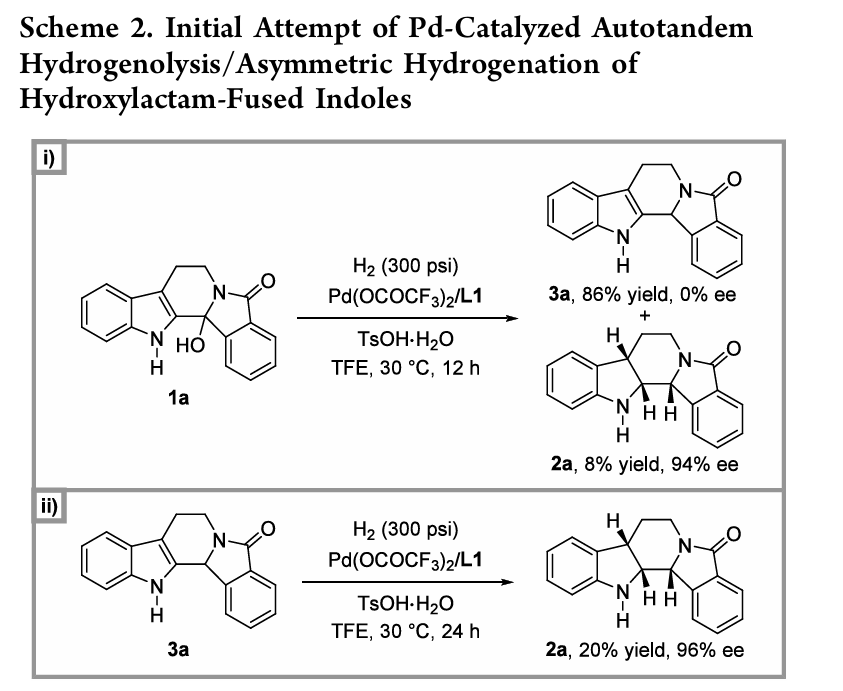
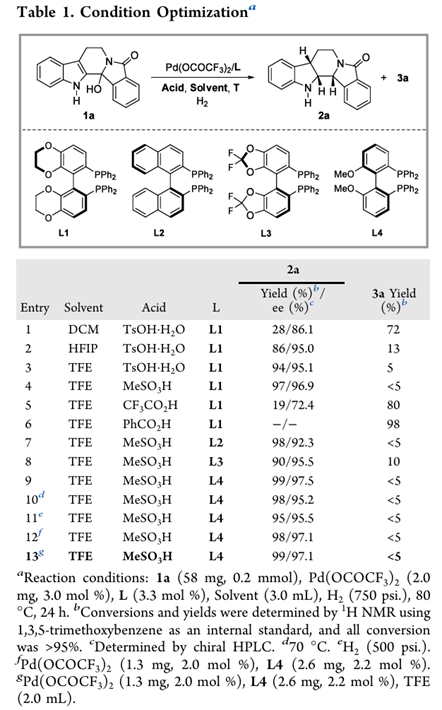
1、反应发展与条件优化
作者以羟基内酰胺并吲哚为模型底物,建立如下体系:
催化剂:Pd(OCOCF₃)₂/ 手性双膦配体
最优配体:(S)-MeOBiPhep
酸:MeSO₃H
溶剂:TFE
条件:80 ℃,750 psi H₂
在该条件下:
收率:中等到优秀(最高95%)
对映选择性:~97% ee
👉 说明该自串联体系可以高效运行
2、反应模式
该工作实现了:
✅ 氢解 + 不对称氢化 一锅串联
✅ 单一Pd–H活性物种驱动两个反应
✅ DKR参与,实现立体收敛
最终得到:
👉 六氢β-咔啉(hexahydro-β-carbolines)
👉 含三个连续手性中心
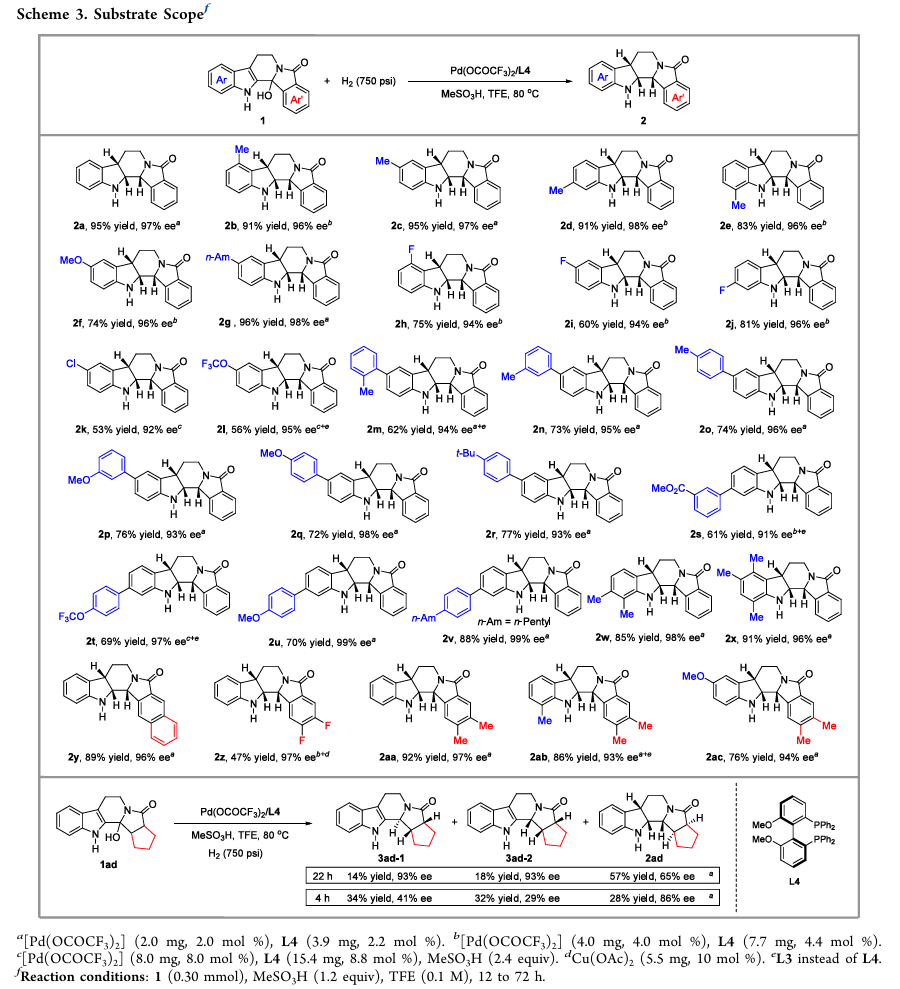
3、底物适用范围(Scope)
该反应具有良好的普适性:
吲哚环:
给电子/吸电子取代均适用
芳基/多取代体系:兼容
羟基内酰胺部分:多种取代可行
结果:
收率:中等到优秀
ee:普遍优秀(>90% ee)
特殊例子:
可构建5个连续手性中心产物(通过DKR)
通过延长/缩短反应时间还可分离得到KR产物3ad-1/3ad-2
4、合成应用(Synthetic Utility)
该方法具有良好的实用性:
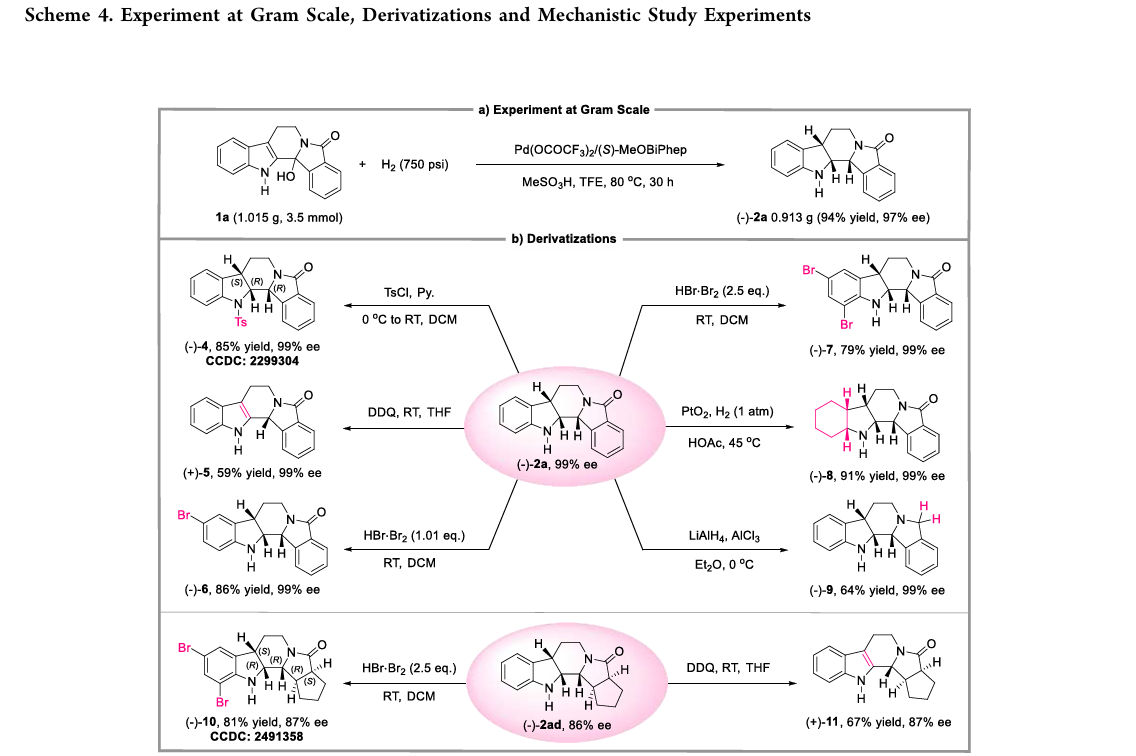
✅克级反应:94% yield,97% ee
✅ee可通过重结晶提升至99%
✅可进一步转化为:
N-Ts衍生物
芳构化产物
溴化衍生物
多环结构
👉 且所有转化不损失对映纯度
机理解析
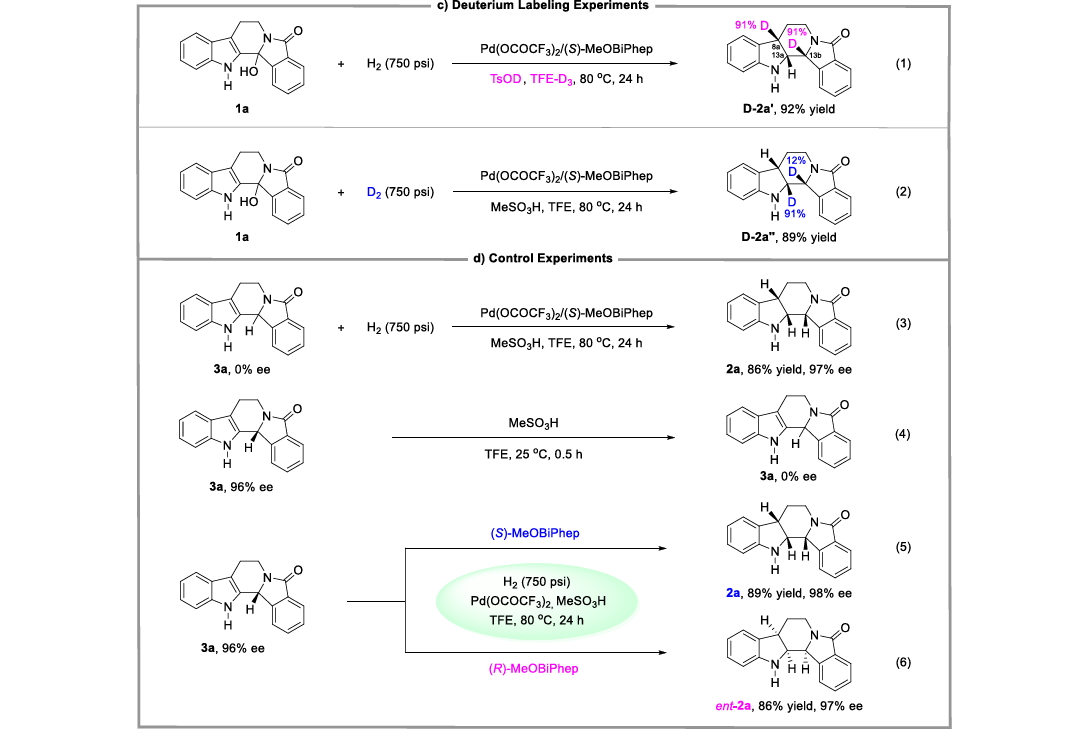
🔬 关键实验结论
(1)DKR过程存在
氘代实验表明:
存在快速可逆的质子化/去质子化过程
(2)中间体会消旋
氢解产物在酸作用下:
👉快速消旋(racemization)
(3)对映选择性来源
最终ee:
👉完全由手性Pd催化剂控制,而非对映选择性则由DKR过程共同控制(快速消旋+选择性氢化)
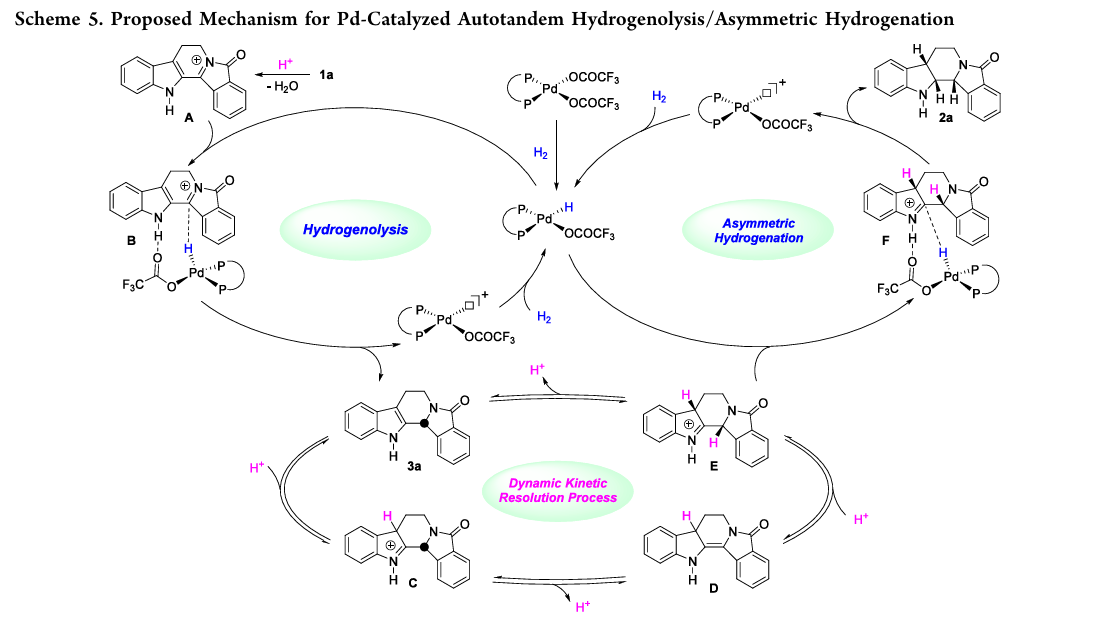
整个反应过程可以概括为:
氢解生成中间体(外消旋)
在酸作用下发生快速互变(消旋)
形成亚胺盐中间体
Pd–H进行不对称氢化
优势构型被选择性放大(DKR)
👉 DKR是实现高对映与高非对映选择性的核心
总结
该工作发展了一种全新的钯催化不对称自串联反应体系,利用单一Pd–H活性物种同时驱动C–O键氢解与吲哚不对称氢化两步反应,借助动态动力学拆分(DKR)过程实现立体收敛,高效构建了含有三个连续立体中心的六氢β-咔啉化合物,甚至可进一步扩展至含有五个连续手性中心的八氢吲哚衍生物。该方法展现出高收率、高对映选择性(ee)和高非对映选择性(dr)、优异的底物普适性以及强大的合成应用潜力。本工作为金属氢化物(Pd–H)驱动的不对称自串联催化提供了首个氢解/氢化串联范例,并为复杂多手性中心骨架的一步高效构建开辟了新策略。
文章链接:
https://doi.org/10.1021/jacs.5c20093


