

 1406
1406
键解离能是指将分子中的一个化学键断裂成两个自由基所需的能量,通常以kcal/mol或kJ/mol为单位。它反映了键的稳定性:BDE越高,键越难断裂。
在实际应用中,BDE常用于:
预测反应活性(如自由基反应)。
评估抗氧化剂的效能(例如,酚类化合物的O-H键BDE)。
设计新型材料(如高分子聚合物的稳定性)。
传统实验测量BDE耗时费力,而计算方法如Gaussian可以快速提供可靠数据。当然,计算结果的准确性取决于方法和基组的选择。
Gaussian支持从HF(Hartree-Fock)到高级的CCSD(T)等方法。它可以处理分子优化、能量计算、振动频率等任务。最新版本如Gaussian 16(G16)优化了计算效率,支持并行计算。
如果你是初学者,建议从Gaussian 09W或G16入手。安装后,通过输入文件(.gjf或.com格式)提交任务,输出文件(.log)会给出结果。Gaussian不是免费软件,但许多大学有授权。
计算键解离能的基本原理
BDE的计算通常基于同核解离(homolytic cleavage),公式为:
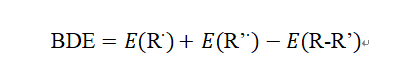
其中:
是完整分子的能量。
和
是断键后自由基的能量。
注意:自由基是开壳层体系,需要使用无限制方法(如UB3LYP)来处理自旋污染。
为了得到更准确的焓值(Bond Dissociation Enthalpy,BDE),需加上零点能(ZPE)和热校正(从0 K到298 K):
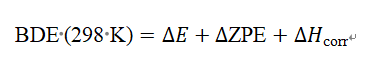
这些可以通过频率计算获得。
计算流程
以下是计算BDE的标准步骤,以甲烷(CH₄)的C-H键为例。方法选择B3LYP/6-31G(d),这是入门级的DFT方法,平衡了准确性和计算成本。
步骤1:分子几何优化
首先优化完整分子的几何结构。
输入文件示例(ch4_opt.gjf):
text
复制
%chk=ch4_opt.chk
#p opt b3lyp/6-31g(d) freq
CH4 optimization
0 1
C
H 1 1.09
H 1 1.09 2 109.47
H 1 1.09 2 109.47 3 120.0
H 1 1.09 2 109.47 3 -120.0
%chk:保存检查点文件。
#p opt:优化几何,freq计算频率(用于ZPE)。
电荷和自旋多重度:0 1(闭壳层)。
分子坐标:用笛卡尔或Z-matrix格式。
运行后,从.log文件中提取优化能量和ZPE。
步骤2:自由基碎片优化
对于C-H键断裂,得到CH₃• 和 H•。
CH₃• 输入文件(ch3_rad_opt.gjf):
text
复制
%chk=ch3_rad_opt.chk
#p opt ub3lyp/6-31g(d) freq
CH3 radical optimization
0 2
C
H 1 1.09
H 1 1.09 2 120.0
H 1 1.09 2 120.0 3 120.0
自旋多重度:2(双重态)。
使用u前缀表示无限制计算。
H• 输入文件(h_rad_opt.gjf):
text
复制
%chk=h_rad_opt.chk
#p opt ub3lyp/6-31g(d) freq
H radical optimization
0 2
H
H•是单电子体系,也需优化(虽然简单)。
步骤3:能量计算与校正
从每个.log文件提取:
SCF能量(电子能量)。
ZPE(零点能,从频率计算)。
热校正到焓(Thermal correction to Enthalpy)。
计算BDE:
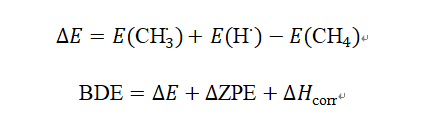
对于CH₄,实验BDE约105 kcal/mol,B3LYP计算值接近这个。
步骤4:可选高级计算
单点能量:优化后用更大基组(如cc-pVTZ)计算能量,提高准确性。关键词:#p sp。
BSSE校正:用反向极化校正(Counterpoise)处理基组叠加误差。关键词:counterpoise=2。
溶剂效应:用PCM或SMD模型模拟溶剂。关键词:scrf=(smd,solvent=water)。
注意事项与常见坑
方法选择:DFT(如B3LYP、M06-2X)适合大分子,波函数方法(如CCSD(T))更准确但计算量大。
基组:从小基组优化(如6-31G(d)),大基组单点(如def2-TZVP)。
自旋污染:检查<S²>值,应接近75(双重态)。如果>0.8,用PUHF或ROHF校正。
收敛问题:如果优化失败,试试opt=(calcfc,tight)或改变初始猜想。
单位转换:Gaussian输出以Hartree,1 Hartree = 627.509 kcal/mol。
示例结果解读
以CH₄为例,计算BDE约为104.5 kcal/mol,接近实验值104.9 kcal/mol。误差来源主要是方法局限性。
总结
使用Gaussian计算键解离能的核心流程是:先对完整分子和两个自由基碎片分别进行几何优化+频率计算,提取电子能量、零点能及298 K热焓校正后,按照BDE = [E(自由基1) + E(自由基2) – E(分子)] + ΔZPE + ΔH_corr公式即可得到结果。选择合适的DFT泛函(如B3LYP、M06-2X)和基组、注意自由基自旋处理与收敛问题,就能获得与实验值相当接近的可靠数据,为你的化学研究提供强有力的量化支持。


