

 2525
2525
d带中心理论由Nørskov等人在20世纪90年代基于密度泛函理论(DFT)提出,是现代表面催化学科的奠基性框架。它将过渡金属催化剂的电子结构(d轨道电子分布)与宏观吸附行为、催化活性直接定量关联,为从试错法向理性设计转变提供了核心工具。该理论的核心洞见在于:催化剂表面吸附能并非孤立现象,而是由d带中心(εd)这一电子描述符精准调控的εd位置决定了金属d轨道与吸附质轨道间的杂化强度,从而决定反应中间体的结合强度。吸附过强会导致表面中毒或脱附困难,吸附过弱则无法有效活化反应物;d带中心正是实现适中吸附的电子开关,帮助我们理解Sabatier原理的微观本质,并在HER、ORR、CO₂还原等清洁能源反应中指导高效催化剂设计。
d带中心(εd)的精确定义是d轨道态密度ρd(E)的能量加权平均值,数学表达式为:
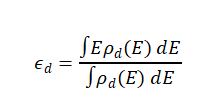
其中积分范围通常取费米能级E_F以下的d带区域,εd相对于EF的位置是关键。当εd更靠近EF(上移)时,未占据d轨道增多,反键轨道填充度降低,吸附键增强;反之,下移则反键轨道被更多电子占据,吸附削弱。这一公式通过DFT计算的投影态密度(PDOS)即可精确获得,为实验表征(如XPS、UPS)提供了理论锚点。吸附能(ΔE_ads)则定义为吸附前后体系总能之差(负值表示放热吸附),二者呈现强线性相关:ΔE_ads ∝ ε_d(R² > 0.8)。
(https://www.nature.com/articles/s41524-022-00846-z)
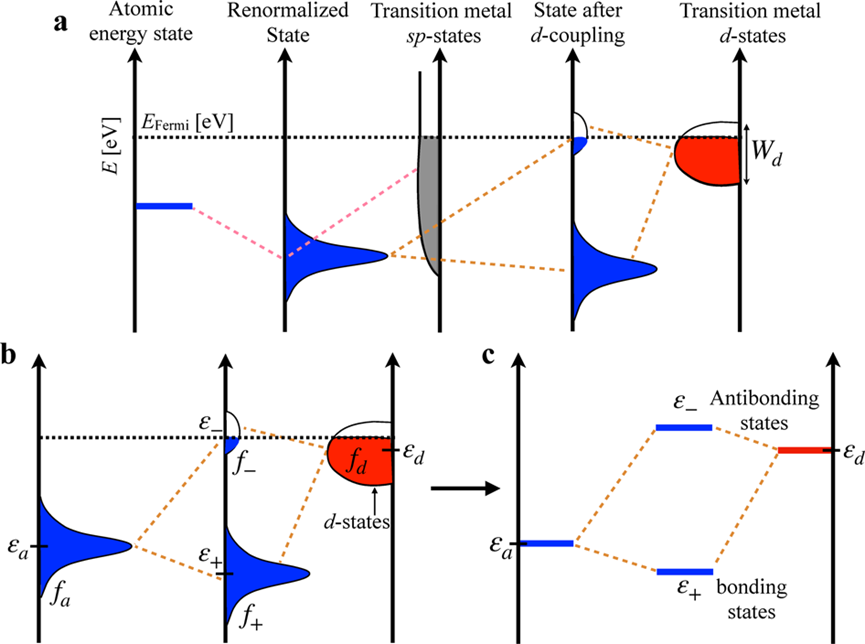
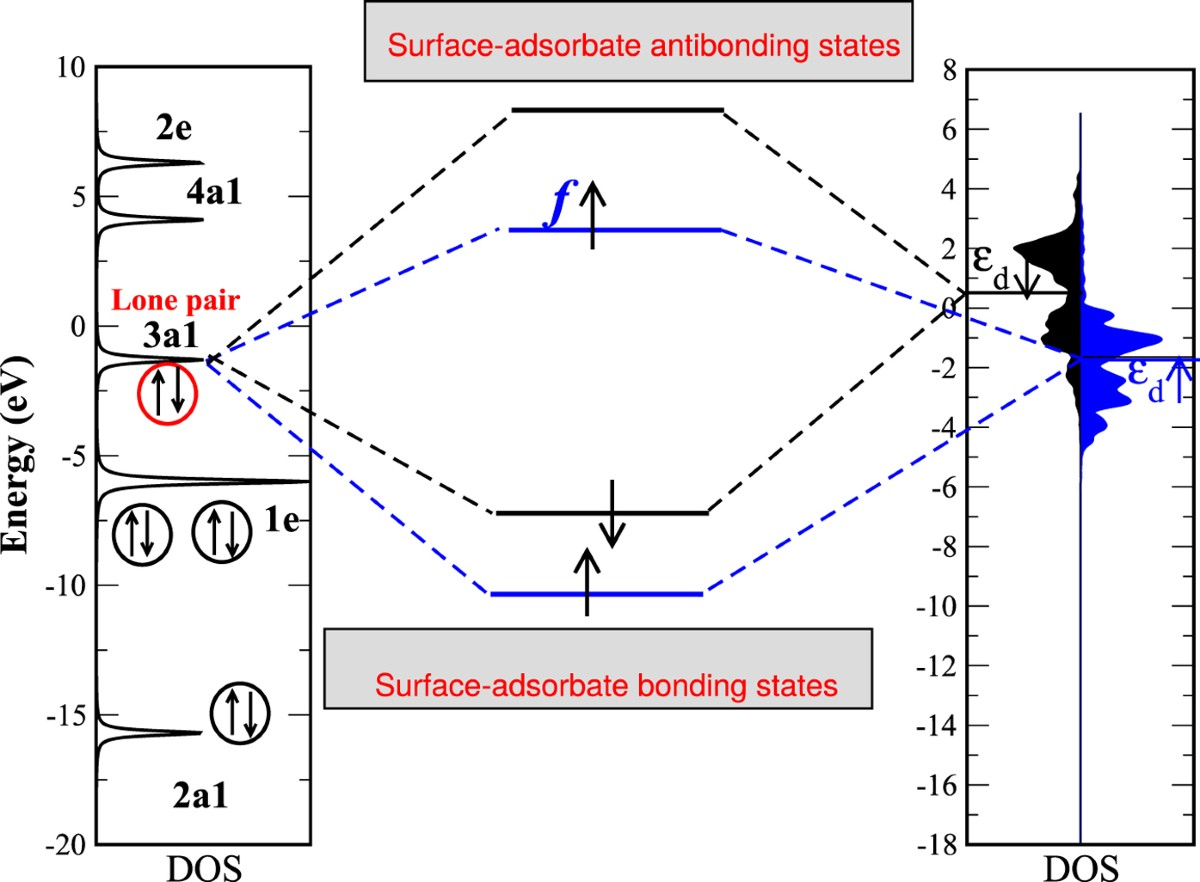
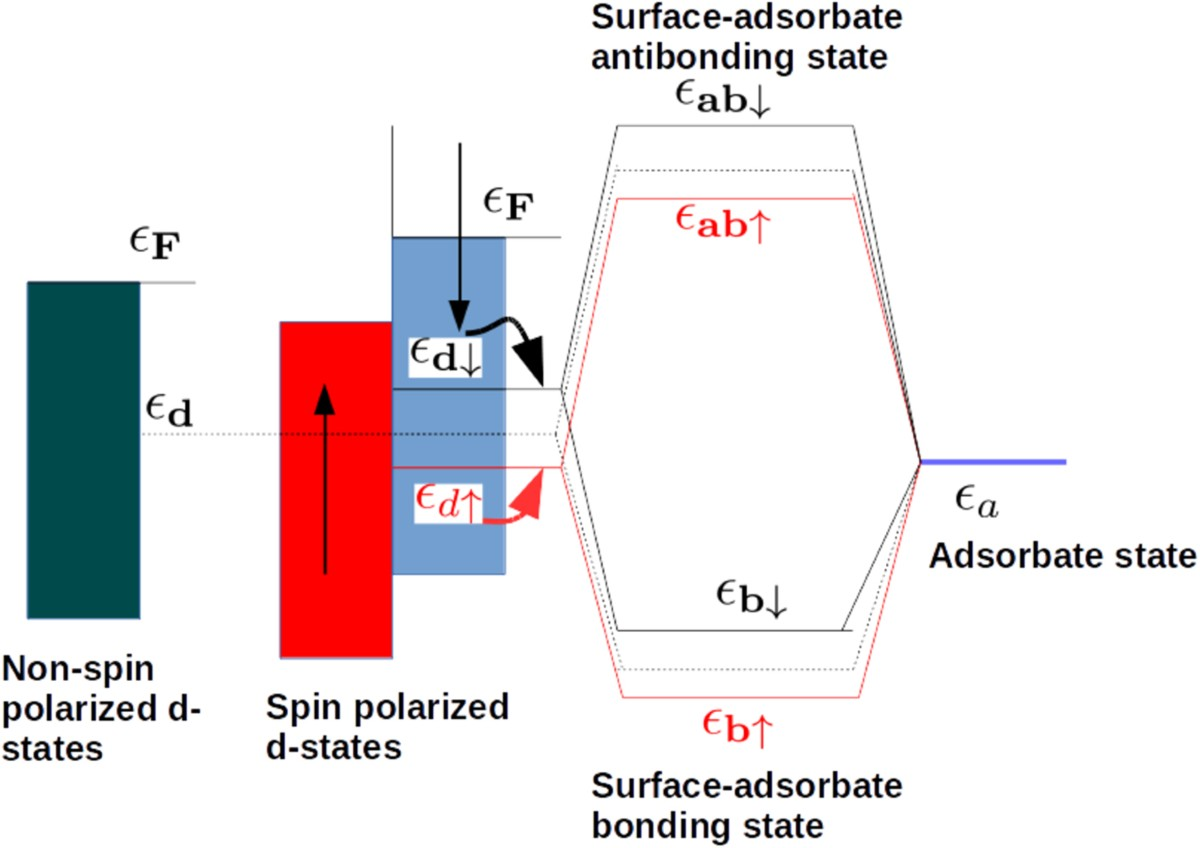
d带中心调控吸附能的机制源于d轨道与吸附质价轨道的轨道杂化。吸附物(如O₂的π轨道或CO的2π轨道)接近金属表面时,与金属d轨道形成成键态(低能级)和反键态(高能级)。若反键态位于费米能级以下并被电子占据,则成键被削弱,吸附能较弱;若反键态在费米能级以上未被占据,则成键增强,吸附能更负。这一过程完全由εd位置决定:εd上移使反键态上移、更难填充,导致吸附强化;εd下移则相反。这一杂化模型不仅解释了Sabatier原理的电子起源,还揭示了为什么Pt等贵金属在火山曲线峰值附近表现出色其εd恰好平衡活化与脱附。
(https://www.nature.com/articles/srep35916)
催化活性随吸附能(或εd)呈现经典火山曲线(Volcano Plot),这是d带理论最直观的预测工具。火山左侧为强吸附区(εd过高),中间体难以脱附导致活性下降;右侧为弱吸附区(εd过低),反应物活化不足;峰值对应适中吸附能,活性最高。这一曲线直接源于Sabatier原理,但d带中心将其电子结构化:活性A ≈ -|ε_d - ε_opt|(ε_opt为最优位置)。DFT结合PDOS可精确绘制火山图,帮助快速筛选催化剂,避免盲目实验。在ORR中,火山峰对应ΔG_OH* ≈ 0.8-1.0 eV的中间体吸附;HER中则对应H*吸附自由能接近零。火山图的强大之处在于,通过少数关键吸附能即可预测整个反应路径的活性趋势,大幅加速催化剂优化。
d带中心的精准调控是理论走向实践的关键,其上移或下移源于多种微观机制。上移(增强吸附)的原因包括:拉伸应变(d带变窄)、低配位数原子(缺陷、边缘、台阶处d轨道重叠减弱)、电负性差异的电子抽取效应、氧空位等缺陷工程以及界面电场诱导的电子转移。下移(减弱吸附)则对应压缩应变(d带变宽)、高配位数、相转变(如CoSe₂从立方到正交相)、强金属-载体相互作用(电子向载体转移)以及B、N等轻元素掺杂的p-d杂化。这些调控手段已在合金化(Vegard定律近似:ε_d,alloy ≈ x·ε_d,A + (1-x)·ε_d,B)、应变工程、单原子催化剂配位设计中广泛验证。例如,Ni-Co₃O₄通过氧空位上移ε_d增强H*吸附;Pt/WO₃通过载体相互作用下移ε_d优化脱附平衡。深刻理解这些机制,能实现从“被动筛选”到“主动设计”的跨越。
(https://www.nature.com/articles/srep35916)
实际应用中,d带中心理论已催生诸多突破。以HER为例,Pt的εd接近最优,但Ni因εd过高导致H吸附过强;通过PtM合金或应变调控下移εd,可将活性提升至媲美Pt。ORR中,PtFe有序合金通过压缩d轨道上移εd,优化OOH/OH*脱附,半波电位提升超50 mV。CO氧化中,Au/FeOₓ界面电子转移下移Au的εd,打破Au对CO的惰性,实现低温高效氧化。CO₂还原中,PtCu合金通过Cu掺杂精细调控εd,避免碳中毒,同时提升C₂产物选择性。这些案例证明:d带中心不仅是描述符,更是分子级开关,结合机器学习和原位谱学(如XAS),可预测复杂合金的局部εd,进一步推动非贵金属、单原子催化剂的工业化。
(https://www.nature.com/articles/s41467-024-50377-y)
尽管d带中心理论强大,但仍存在局限:在强关联体系(如氧化物)、溶剂化环境或动态表面重构下,静态DFT计算可能偏差;标度关系也存在“破火山”现象。未来,通过原位表征、机器学习加速筛选以及多尺度模拟,该理论将与AI催化剂设计深度融合,推动绿色氢能、碳中和等领域的革命性进展。d带中心理论真正架起了电子结构与宏观性能的桥梁,让催化剂设计从走向科学。


