

 251
251

一、研究背景与问题
八元氮杂环骨架广泛存在于天然生物碱及人工合成化合物中具有蛋白激酶抑制、抗癌、麻醉受体相互作用和杀虫等多种生物活性。此类稠合多环衍生物还因其独特的三维刚性结构可用作配体、功能染料、荧光探针、半导体及光伏材料。然而,中等环尺寸的氮杂环的构建因不利的熵效应和焓应变而极具挑战性。
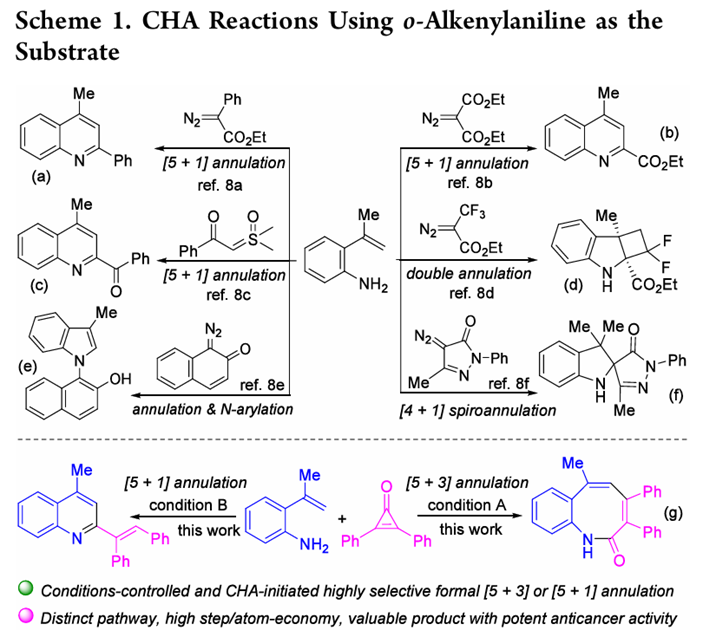
另一方面,功能化喹啉是最重要的氮杂环化合物之一。其中,2-烯基取代喹啉既是多功能合成中间体又具有抗疟、抗利什曼病、哮喘抑制及CysLT1拮抗等广泛药理活性。尽管已有Wittig反应、2-甲基喹啉与醛/亚胺的缩合以及喹啉N-氧化物直接乙烯化等合成策略,但开发更简洁、高效、绿色且通用的方法仍是迫切需求。在C−H键活化领域,使用自由氨基作为导向基具有独特优势:其广泛存在于廉价原料中,配位能力强,且氨基具有显著亲核性,可引发多种后偶联反应。尽管如此,烯基C(sp2)−H键的直接官能化相较于芳基C(sp2)−H键仍然是一个难题。
已有的研究借助2-烯基苯胺底物利用氨基作为自由基实现烯基C(sp2)−H键官能化但已报道的工作大多聚焦于重氮化合物或亚砜鎓叶立德作为偶联伴侣,主要生成六元或五元氮杂环骨架。将偶联伴侣从重氮化合物拓展至环丙烯酮并探索不同条件下选择性合成不同类型杂环产物正是本文致力于解决的核心问题。
二、文章亮点
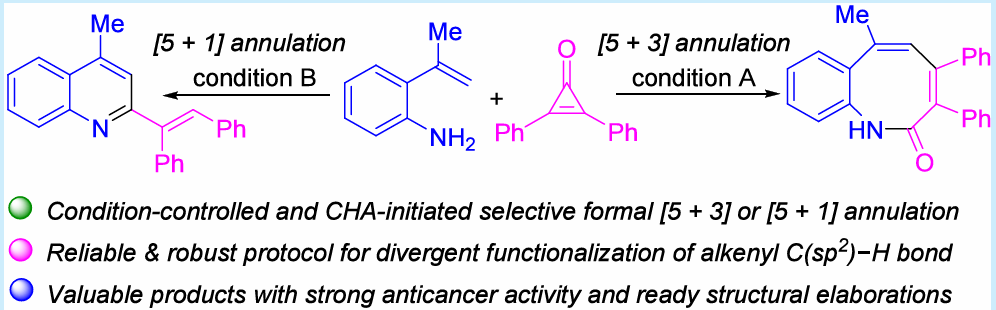
条件可控的选择性:文章仅通过改变反应条件(溶剂、添加剂、温度),即可从相同起始原料精准切换合成路径,选择性地得到苯并[b]氮杂辛酮(product 3,[5+3]环化)或2-烯基喹啉(product 4,[5+1]环化)实现了多样性导向合成。
文章首次将环丙烯酮引入2-烯基苯胺的Rh(III)催化C−H键活化反应体系突破了已有工作对重氮化合物/亚砜鎓叶立德的依赖,安全性更优。
文章实验方法具有优异的原子经济性,反应放大至3 mmol规模仍能以中等至良好收率获得目标产物(3a:53%,64 g;4a:40%,0.46 g)。
苯并[b]氮杂辛酮产物3系列(3a−3hh)和2-烯基喹啉产物4系列(4a−4z)均展现出良好的底物普适性,包容多种供电子/吸电子取代基及不同位置的取代基。
苯并[b]氮杂辛酮衍生物对SW-480、HCT-116、HeLa三种人癌细胞株表现出强抑制活性与阳性对照5-FU相当具有良好的先导化合物开发潜力;而2-烯基喹啉产物4未显示出显著抗增殖活性。
产物可多样化修饰:3a和4a可进一步转化为2-氯苯并[b]氮杂辛、3,4,6-三苯基苯并[b]氮杂辛、2-苯甲酰喹啉及多环产物等体现了良好的结构拓展性。
三、主要内容
反应条件优化
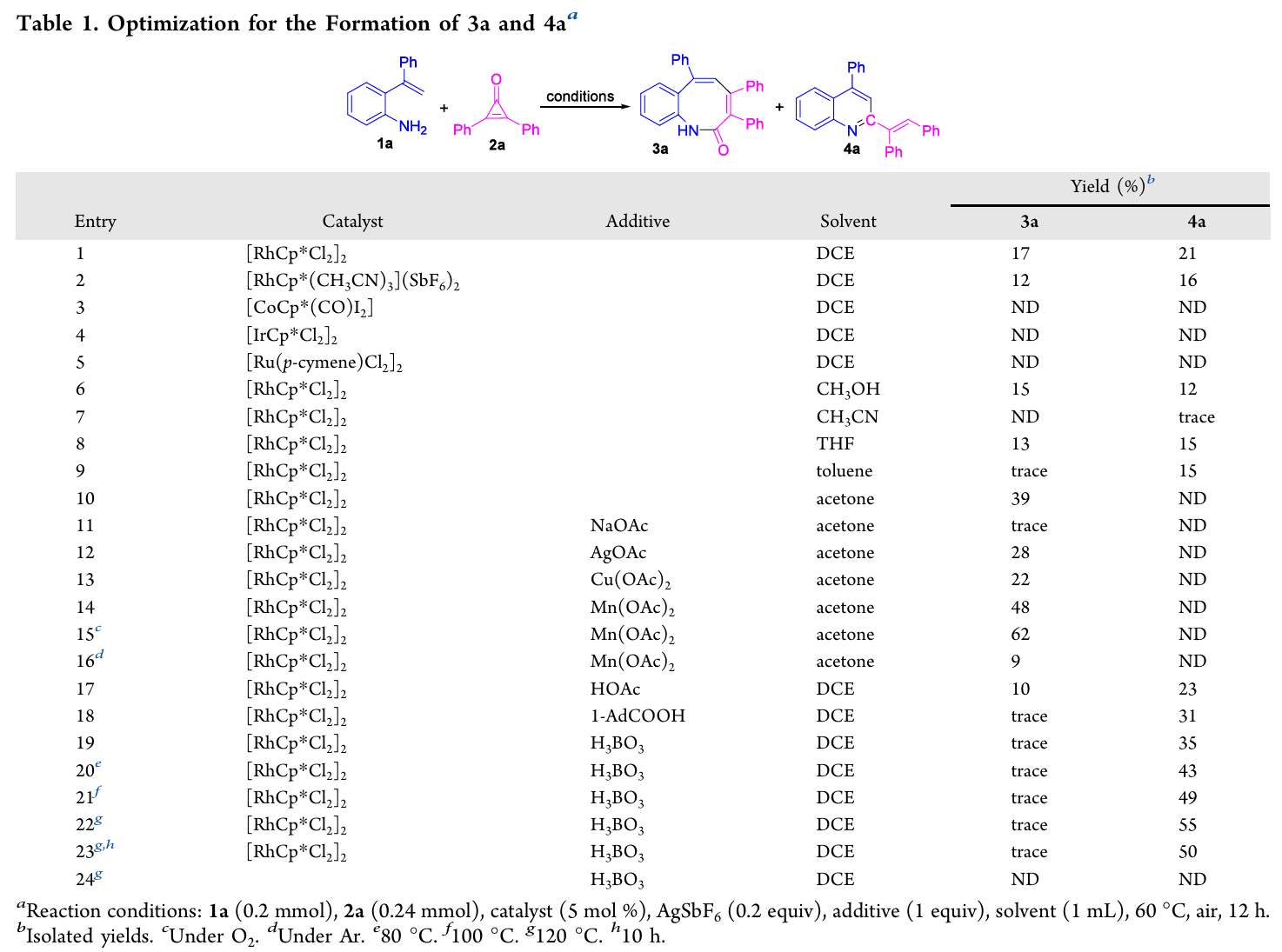
作者以1a与2a为模型底物,[RhCp*Cl2]2/AgSbF6为催化体系系统筛选了催化剂、溶剂、添加剂及反应温度。关键发现如下:
催化剂:[RhCp*Cl2]2效果最优;[CoCp*(CO)I2]、[IrCp*Cl2]2、[Ru(p-cymene)Cl2]2均无效。
合成3的最优条件(条件A):丙酮溶剂 + Mn(OAc)2(1 equiv)+ O2气氛 + 60°C,12 h,3a收率62%,无4a生成。
合成4的最优条件(条件B):DCE溶剂 + H3BO3(1 equiv)+ 空气 + 120°C,12 h,4a收率55%,3a仅痕量。
无催化剂时反应不发生(entry 24)证明了Rh(III)催化必不可少。
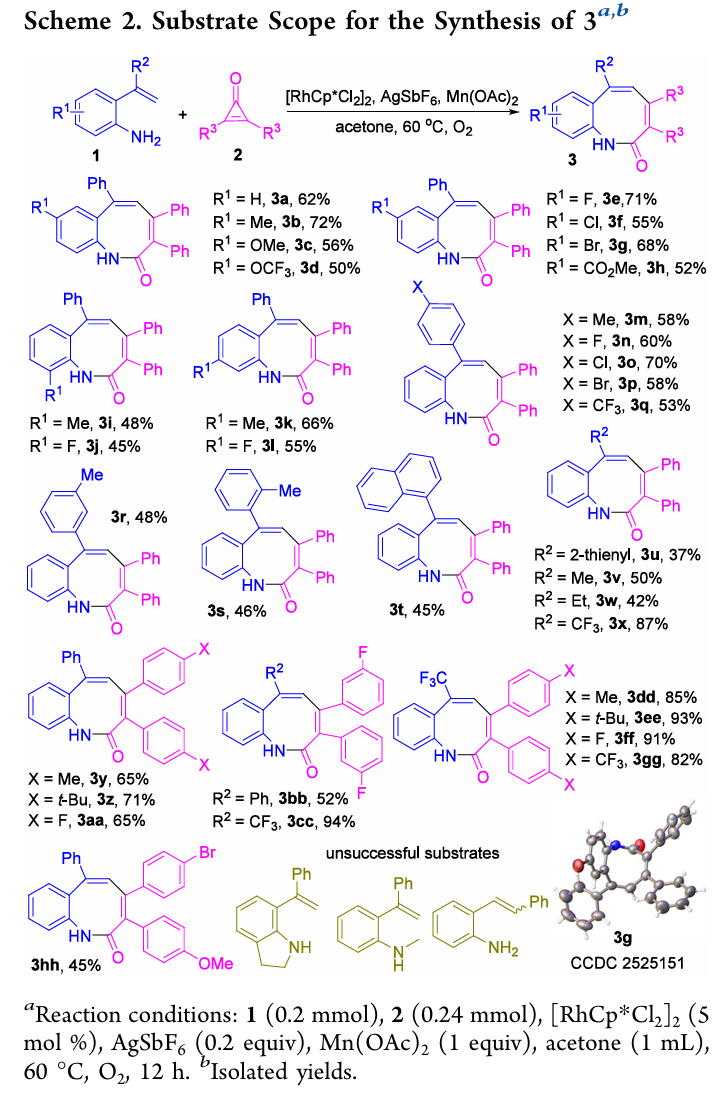
底物适用范围
苯并[b]氮杂辛酮(product 3)系列:苯胺环上对位含供电子基(Me、OMe)或吸电子基(OCF3、F、Cl、Br、酯基)的底物1均可反应,以中等至优秀收率得到3a−3h。邻位(2-Me、2-F)、间位(Me、F)取代底物也同样适用(3i−3l)。R2位为取代苯基、萘基、2-噻吩基及脂肪烷基(Me、Et、CF3)的底物均兼容(3m−3x)。环丙烯酮2的适用范围涵盖EDG(Me、t-Bu)和EWG(F)取代以及不对称环丙烯酮(3hh)。2-(3,3,3-三氟丙-1-烯-2-基)苯胺与环丙烯酮反应收率为82−94%(3cc−3gg)。
2-烯基喹啉(product 4)系列:苯胺环上对位、间位、邻位含Me、OMe、F、Cl、Br、CF3、NO2等取代基的底物均能以[5+1]环化顺利得到4a−4k。特殊底物(苯并二氧戊环胺、萘胺衍生物)及R2含取代苯基或甲基的底物也适用(4l−4t)。2-(环戊-1-烯-1-基)苯胺可生成菲啶衍生物4u。不对称环丙烯酮反应区域选择性给出了单一区域异构体4z。
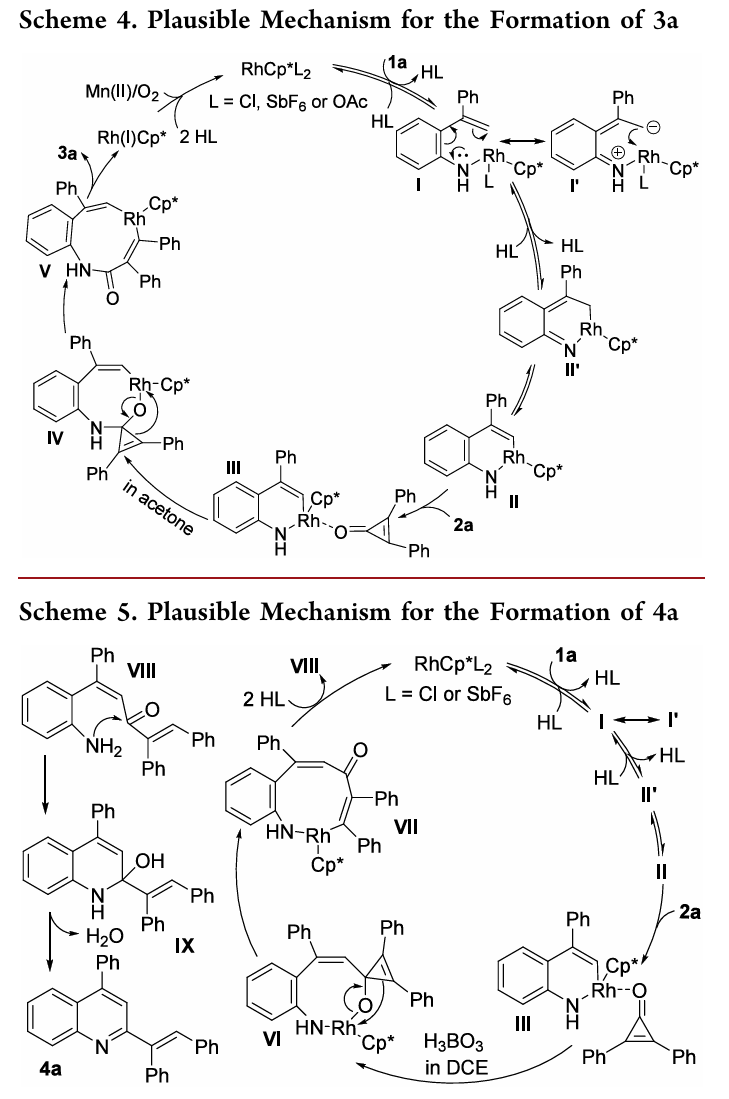
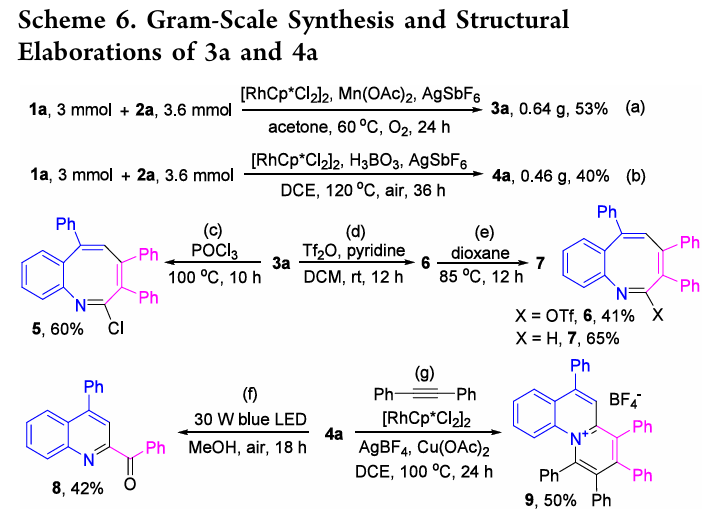
反应机理
基于实验机理研究及文献报道,提出了两条反应路径的合理机制:
合成3a的路径([5+3],条件A/丙酮):Rh(III)与1a配体交换生成中间体I,经分子内C-亲核进攻给出II;II与2a配位后(III),在丙酮中发生2a对N−Rh键的迁移插入得到IV;β-碳消除后经还原消除生成3a,Rh(I)被Mn(OAc)2/O2氧化再生Rh(III)。
合成4a的路径([5+1],条件B/DCE+H3BO3):共享I−III中间体后在DCE/H3BO3条件下发生2a对C−Rh键的迁移插入得VI;β-碳消除后经脱金属质子化得VIII和Rh(III);VIII发生分子内N-亲核加成得IX,最终经芳构化驱动的脱水消除生成4a。
两条路径的分叉点在于中间体III中2a的插入方式:丙酮中优先N−Rh键插入(→3),DCE/H3BO3中优先C−Rh键插入(→4)。
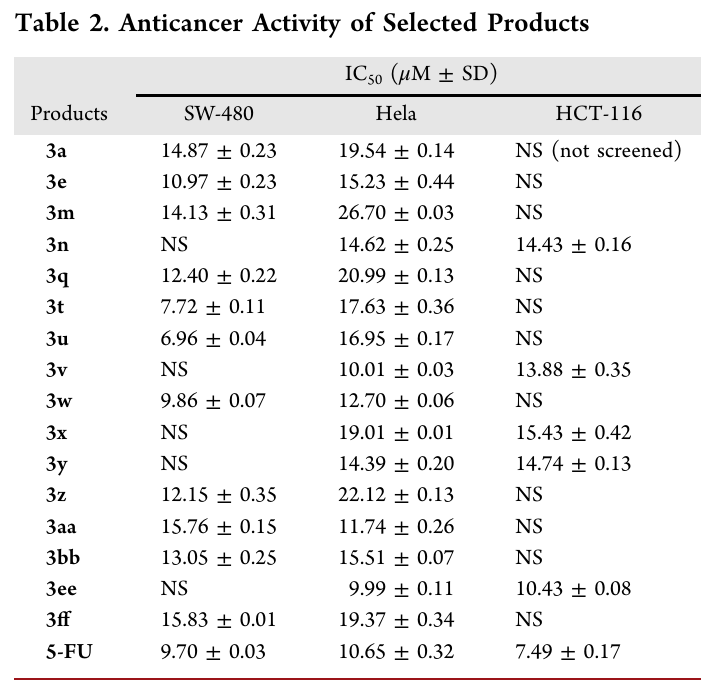
放大合成与产物修饰
3 mmol规模放大合成:3a收率53%(0.64 g),4a收率40%(0.46 g)。产物可以进一步转化:3a经POCl3得2-氯苯并[b]氮杂辛5;经Tf2O/吡啶及热消除得3,4,6-三苯基苯并[b]氮杂辛7;4a经蓝光氧化裂解得2-苯甲酰喹啉8;经Rh(III)催化与二苯乙炔反应得多环产物9。
抗癌活性
作者以5-FU为阳性对照,对三种人癌细胞株(SW-480、HCT-116、HeLa)进行评价。大多数苯并[b]氮杂辛酮衍生物表现出显著抑制活性,IC50值与5-FU接近具有进一步研究潜力。代表性数据:3u对SW-480的IC50为6.96 μM(5-FU:9.70 μM);3ee对HeLa和HCT-116的IC50分别为9.99 μM和10.43 μM(5-FU:10.65 μM和7.49 μM)。2-烯基喹啉产物4在本研究的细胞毒性检测中未显示出显著活性。
总结
本文发展了一种条件可控的选择性合成策略,通过C−H键活化引发的形式[5+3]或[5+1]环化反应,实现了2-烯基苯胺与环丙烯酮的选择性偶联,分别高效合成苯并[b]氮杂辛酮或2-烯基喹啉两类高价值N-杂环骨架。该方法底物易得、反应路径新颖独特,所得苯并[b]氮杂辛酮衍生物具有与阳性对照5-FU相当的显著抗癌活性,且反应原子经济性优异,易于放大,未来在相关领域具有广泛的应用前景。
文章链接
https://doi.org/10.1021/acs.orglett.6c01518


