

 678
678
厦门大学龚磊/林玉妹团队 JACS:利用 Mn(IV)/Mn(III) 双激发配合物实现发散性光诱导不对称转化


在现代合成化学领域,开发高效、可持续且具有高立体选择性的催化体系是核心目标之一。随着光氧化还原催化(Photoredox Catalysis)技术的飞速发展,科学家们已经能够利用可见光驱动许多传统热化学难以实现的转化。然而,长期以来该领域过度依赖于铱(Ir)、钌(Ru)等稀有且昂贵的贵金属光催化剂。
近年来,寻找地壳中丰度更高、毒性更低的 3d 过渡金属替代品已成为绿色化学的前沿阵地。在这一背景下,锰(Mn)作为一种极具潜力的金属元素,逐渐进入了研究者的视野。尽管锰在生物体内(如光合作用中的放氧复合物)发挥着至关重要的光驱动电子转移作用,但在人工光合成及不对称催化领域,其应用潜能尚未得到充分挖掘。
研究背景与挑战
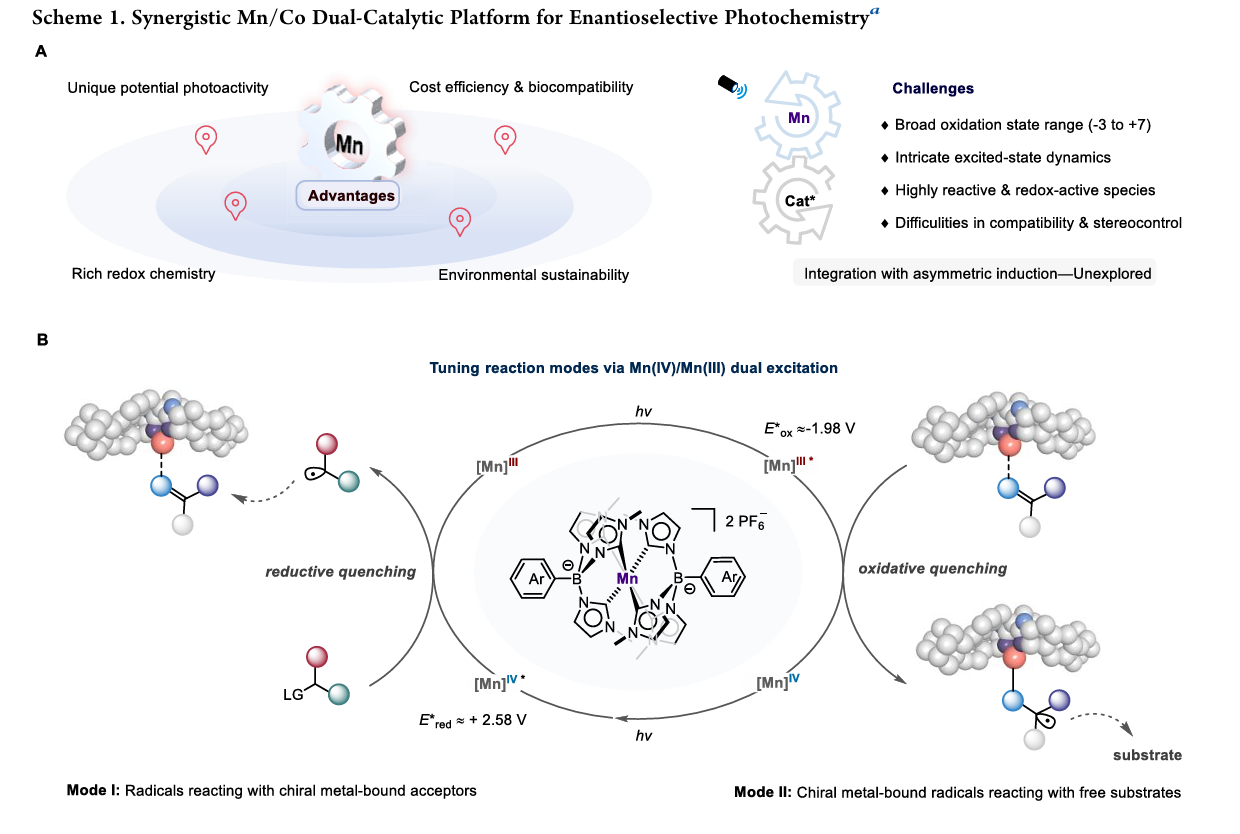
锰催化剂的潜力与局限:锰在地壳中含量丰富、成本低且具有生物兼容性,但在光氧化还原催化领域开发较晚 。其挑战在于锰具有极广的氧化态(-3 到 +7)、复杂的激发态动力学以及极易产生高活性瞬态物种,这使得精确的立体化学控制(不对称诱导)非常困难 。
核心科学问题:如何将锰的光氧化还原能力与手性 3d 过渡金属(如钴)的诱导能力相结合,实现可持续且高对映选择性的转化 。
主要内容
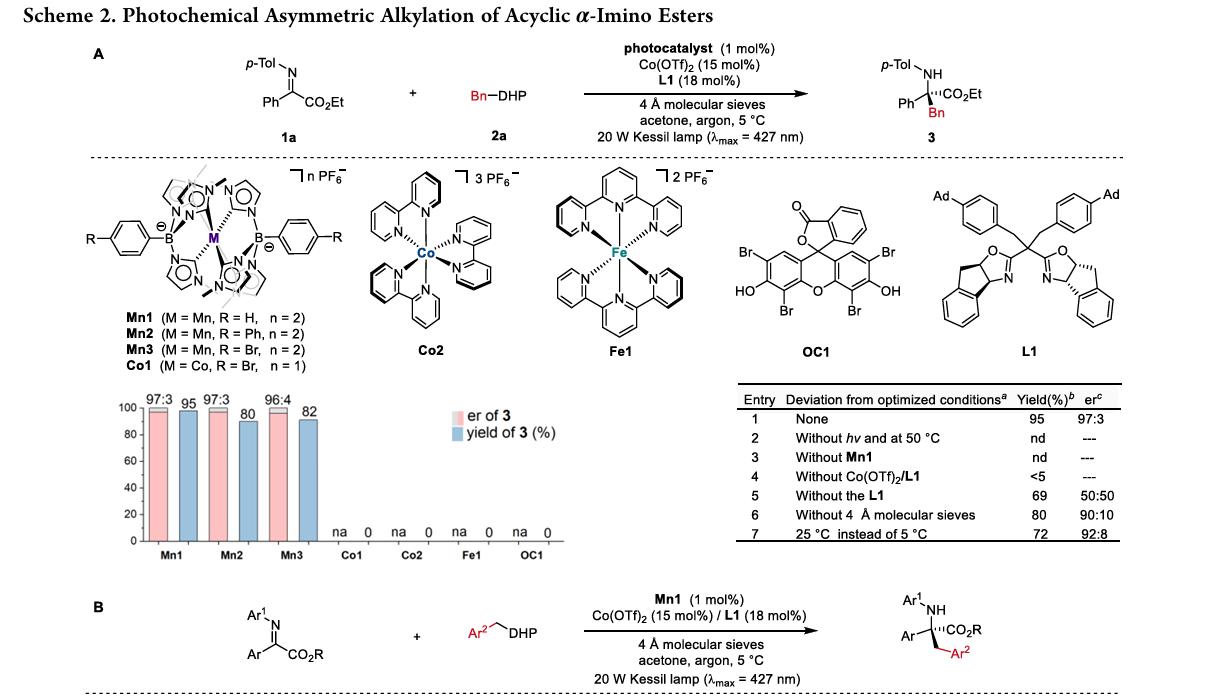
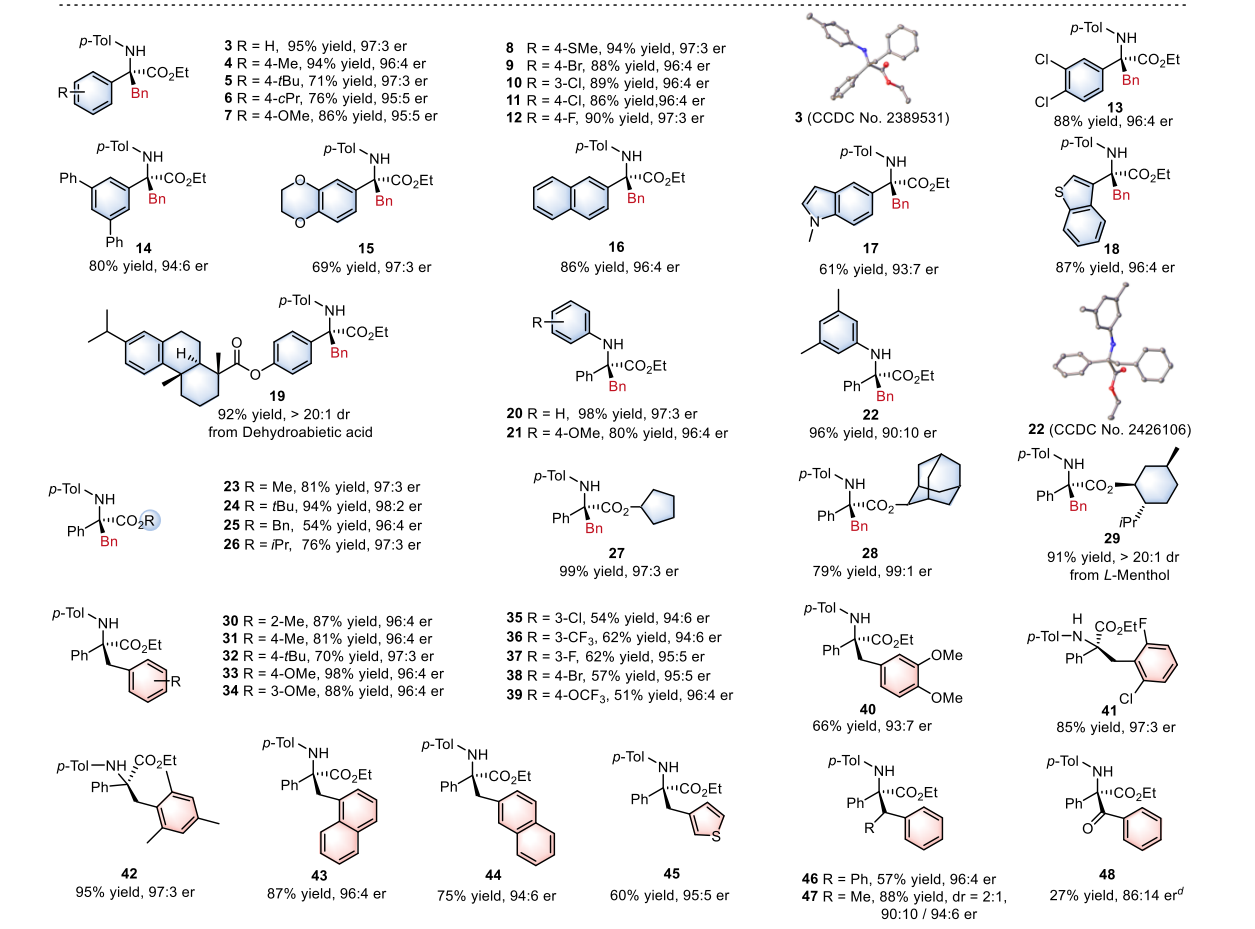
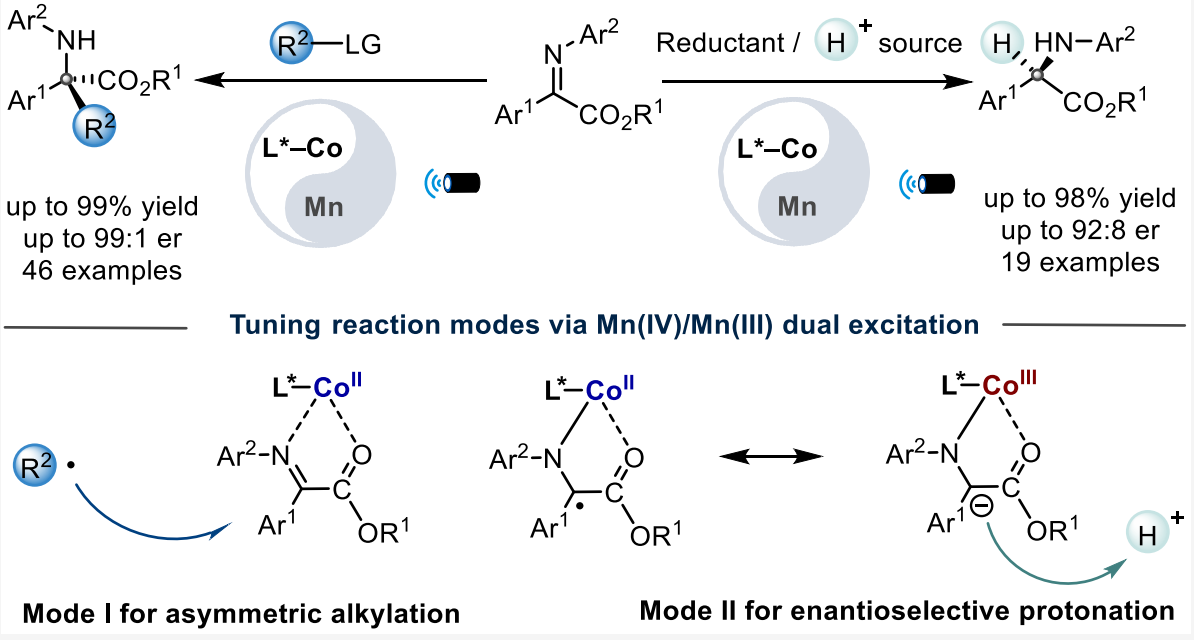
研究团队开发了一种基于 N-杂环卡宾(NHC)配体 的独特锰络合物(Mn1) 。该催化剂具有双激发特性 :
Mn(IV) 激发态:具有极强的氧化能力(E*red ≈ +2.58 V),可氧化自由基前体(如 DHP)产生碳自由基 。
Mn(III) 激发态:具有极强的还原能力(E*ox ≈ -1.98 V),可通过单电子转移还原底物,在手性环境下原位生成金属键合自由基 。
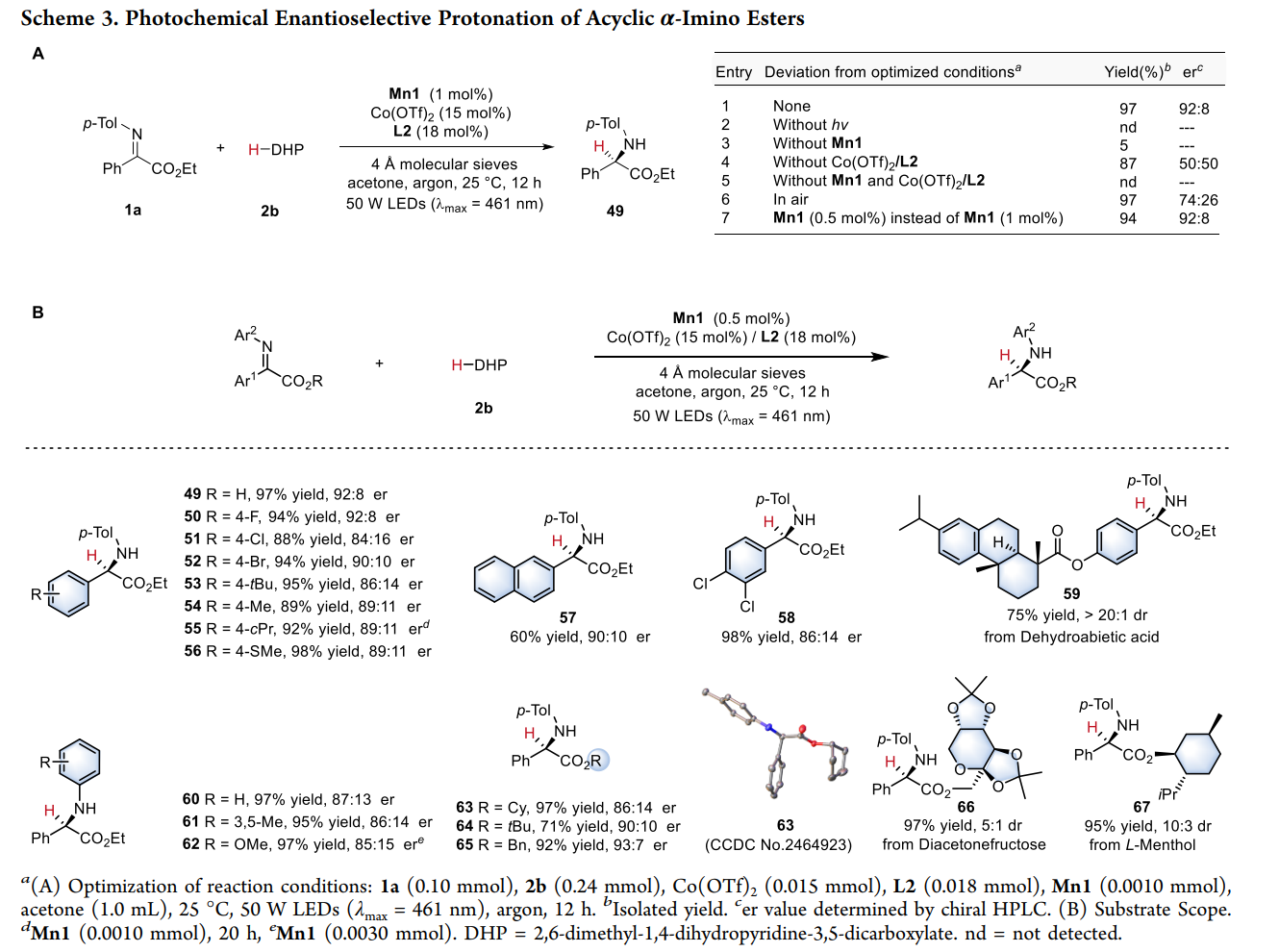
发散性不对称反应模式
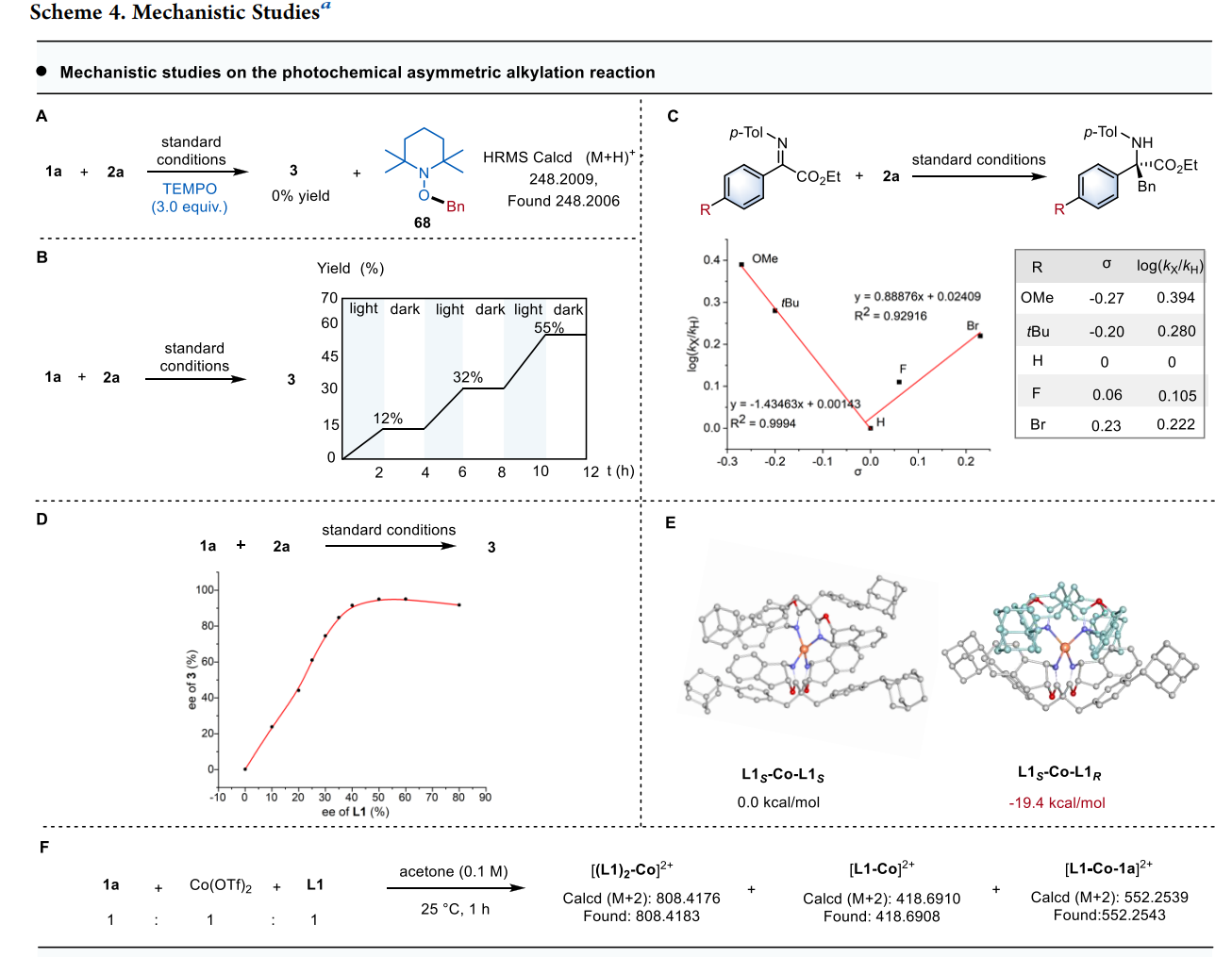
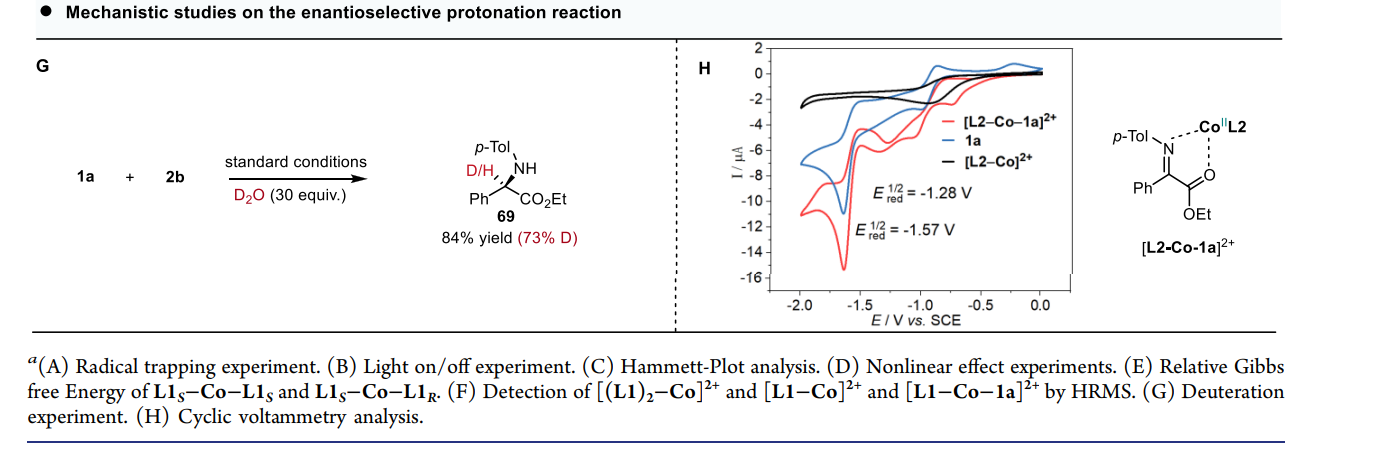
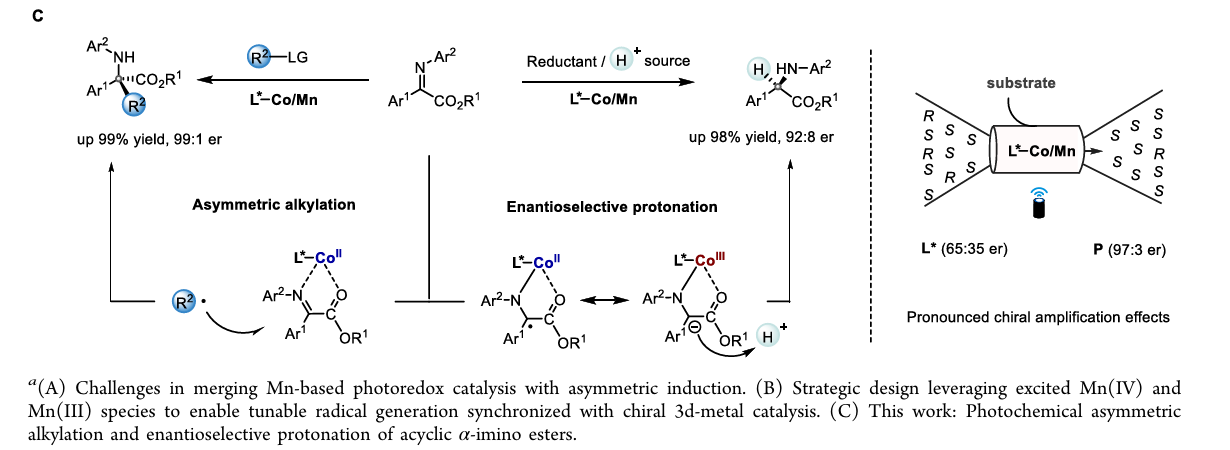
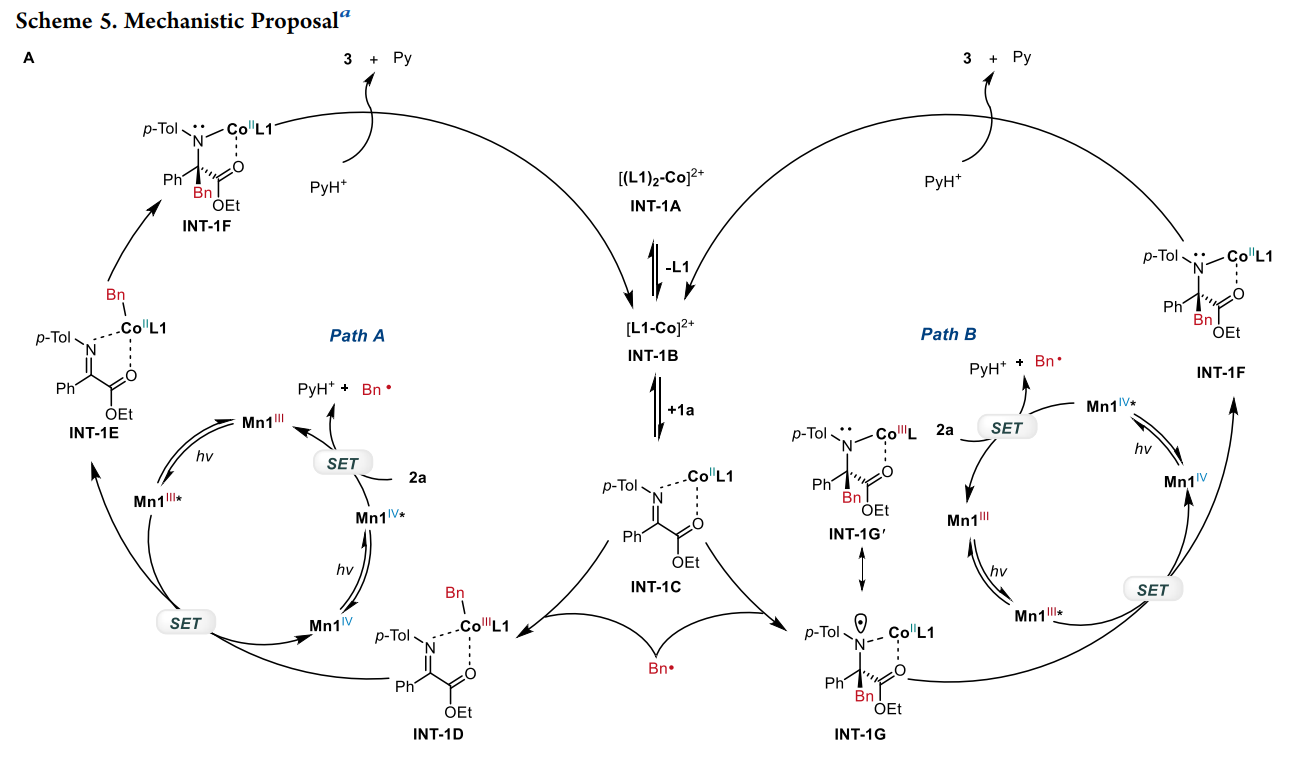
通过这种双激发特性,该系统在同一底物(非循环 α-亚胺酯)上实现了两种完全不同的转化路径 :
模式 I:不对称烷基化(Asymmetric Alkylation)
激发与氧化:Mn(IV) 吸收光子变为 Mn(IV)*,其极高的还原电位(+2.58 V)足以从 Hantzsch 酯衍生物(DHP)中夺取电子,产生烷基自由基 R•。
钴的配位激活:手性 Co(II) 与亚胺底物配位,通过路易斯酸效应降低亚胺的最低未占据轨道(LUMO)能量。
自由基加成:R• 进攻被钴激活的亚胺。由于钴中心周围由手性双噁唑啉(Box)配体构成的手性环境,自由基只能从特定的面进攻,从而决定了季碳中心的手性。
再生:Mn(III) 将电子还给钴结合的中间体,恢复 Mn(IV) 状态并释放产物。
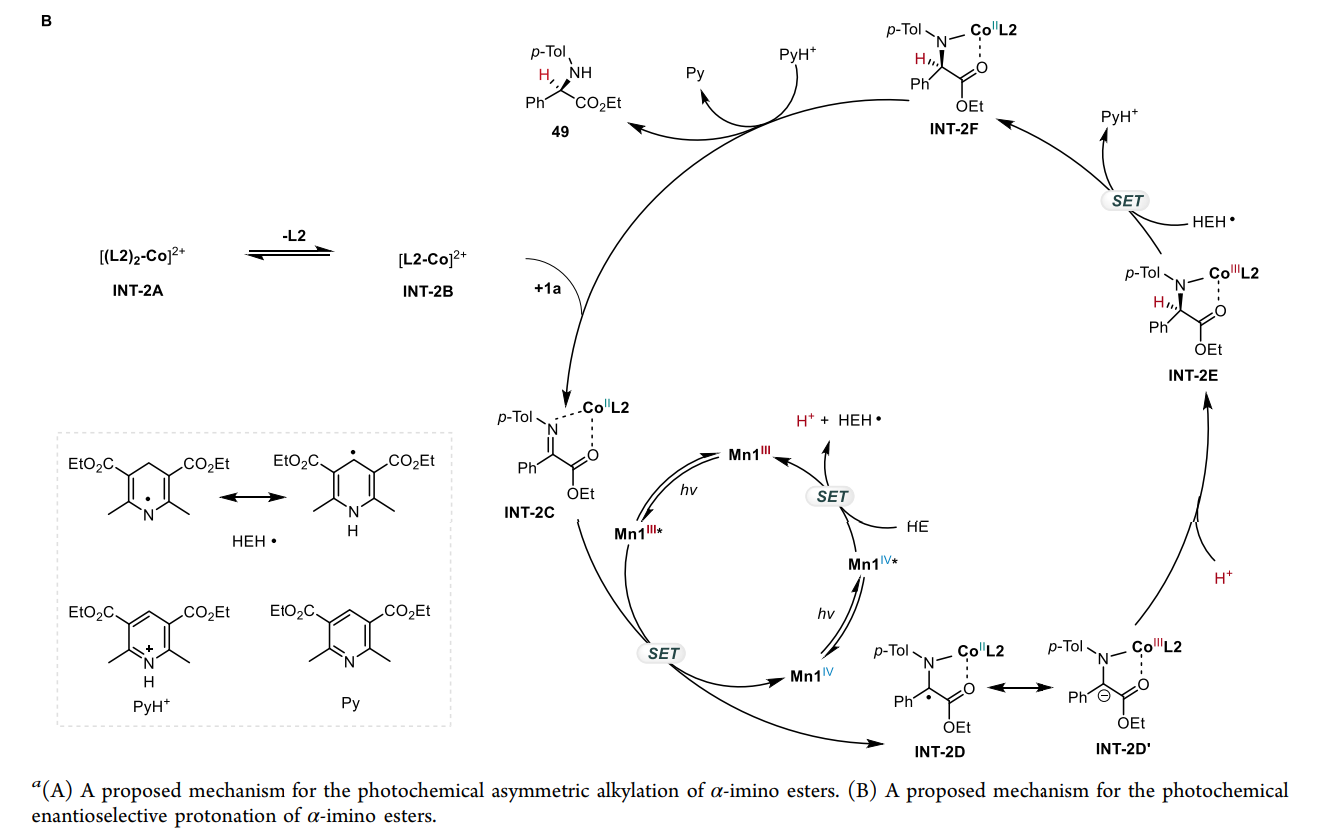
模式 II:对映选择性质子化(Enantioselective Protonation)
直接还原:Mn(III) 吸收光子变为 Mn(III)*,其强还原性(-1.98 V)直接将电子注入到钴结合的亚胺底物中,形成金属结合的自由基阴离子。
受控质子化:这一中间体在手性环境中捕捉质子。质子的来源和加入的方向受手性钴复合物严格限制,从而实现高选择性的质子化,构建叔碳手性中心。
实验发现与重要特性
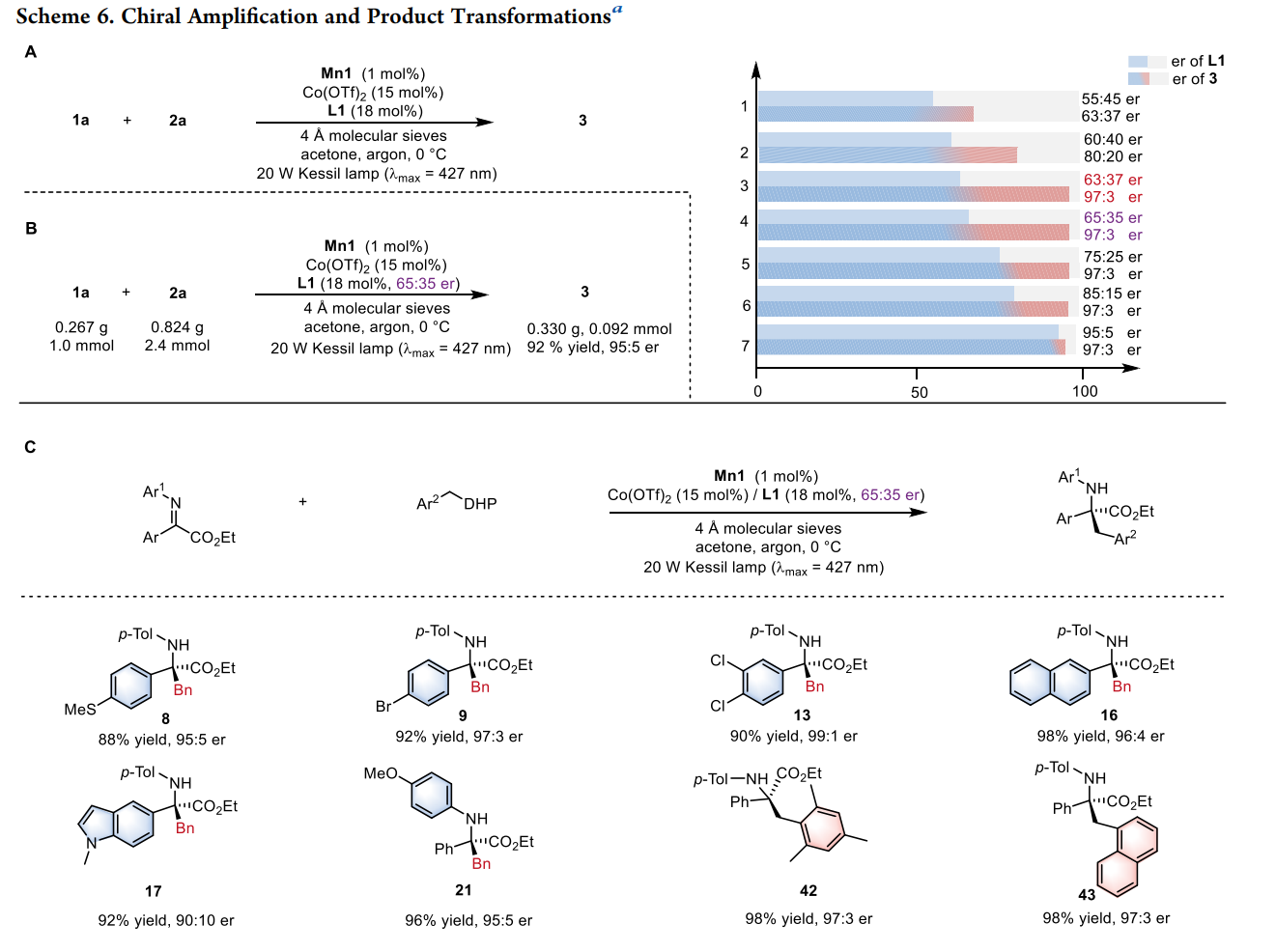
底物普适性:该方法适用于多种芳基、杂芳基及复杂分子骨架(如脱氢枞酸、L-薄荷醇衍生物)。
手性放大效应:系统表现出显著的非线性立体化学放大效应,即使使用低光学纯度的配体,也能得到高对映选择性的产物 。
机械论研究:通过 TEMPO 捕获实验证实了自由基历程 ;光开关实验表明反应严格依赖持续光照 ;HRMS 质谱检测到了关键的钴-底物络合物中间体 。

结论
该研究通过协同使用地球丰度高的锰和钴金属,克服了锰催化中立体选择性控制的难题,为合成高价值手性药物前体和材料科学构建块提供了一个可持续且通用的平台 。


